Resistance: uncommon, most often associated with 23S rRNA G2576T point mutation or acquisition of the cfr ribosomal RNA methyltransferase in staphylococci; clinically related to previous or prolonged drug exposure or horizontal spread
1.2 Tedizolid
NoteFDA-approved indications
Acute bacterial skin and skin structure infections in adults and pediatric patients ≥ 12 years of age caused by susceptible Gram-positive isolates: Staphylococcus aureus (MSSA and MRSA), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus anginosus group, and Enterococcus faecalis.
Usual dose: 200 mg orally or intravenously once daily
Drug interactions: serotonergic and adrenergic interactions possible, but less likely than with linezolid
Serious adverse effects: similar to linezolid but less likely
Resistance: mechanisms similar to linezolid but less common; tedizolid may demonstrate activity against some linezolid-resistant organisms
2 Introduction
The oxazolidinones are a class of antimicrobial agents prepared completely by organic synthesis. In 1978, a patent was issued to E. I. du Pont de Nemours and Company for a series of 5-(halomethyl)-3-aryl-2-oxazolidinones with antimicrobial activity against plant pathogens. Further manipulation of the molecule led to the development of linezolid, which displayed activity against human pathogens (137). A number of oxazolidinones remain investigational; only linezolid (Zyvox) and tedizolid (Sivextro) are currently FDA-approved for clinical use.
3 Chemical Structure
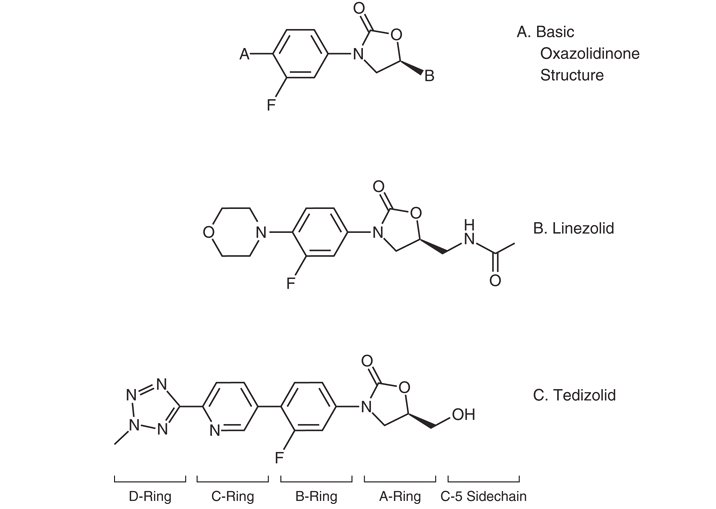
The basic molecular structure of the oxazolidinones is shown in Figure 1 A (126). The structure–function relationships of these compounds have been reviewed (27,58,85,169). Manipulation of the A-ring at the C5 position and manipulation of the N-aryl B-ring are necessary elements of oxazolidinone antibacterial activity. Fluorination of the B-ring further increases activity.
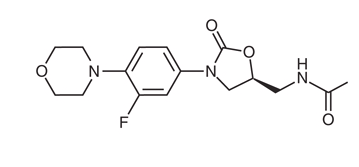
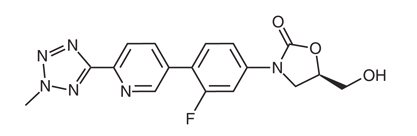
In linezolid (Figure 1 B), the acetamide moiety at the C5 position of the A-ring contributes to overall activity. Tedizolid (Figure 1 C) instead contains a hydroxymethyl group at the same position, which preserves activity against linezolid-resistant organisms carrying the cfr gene. The favorable MIC profile of tedizolid is associated with the pyridine (C-ring) and tetrazole (D-ring) moieties.
Further biochemical manipulations have created new oxazolidinones with potential clinical utility — sutezolid, radezolid, delpazolid, and contezolid — that are presently being evaluated (58).
TipClinical pearl — no cross-resistance
The unique chemical structure of the oxazolidinones makes cross-resistance with β-lactams, vancomycin, quinupristin–dalfopristin, and daptomycin unlikely.
(a) Basic oxazolidinone scaffold
(b) Linezolid
(c) Tedizolid
Figure 1: (A) Basic molecular structure of oxazolidinone antibiotics. (B) Structure of linezolid. (C) Structure of tedizolid. Modified from (126).
4 Mechanism of Action
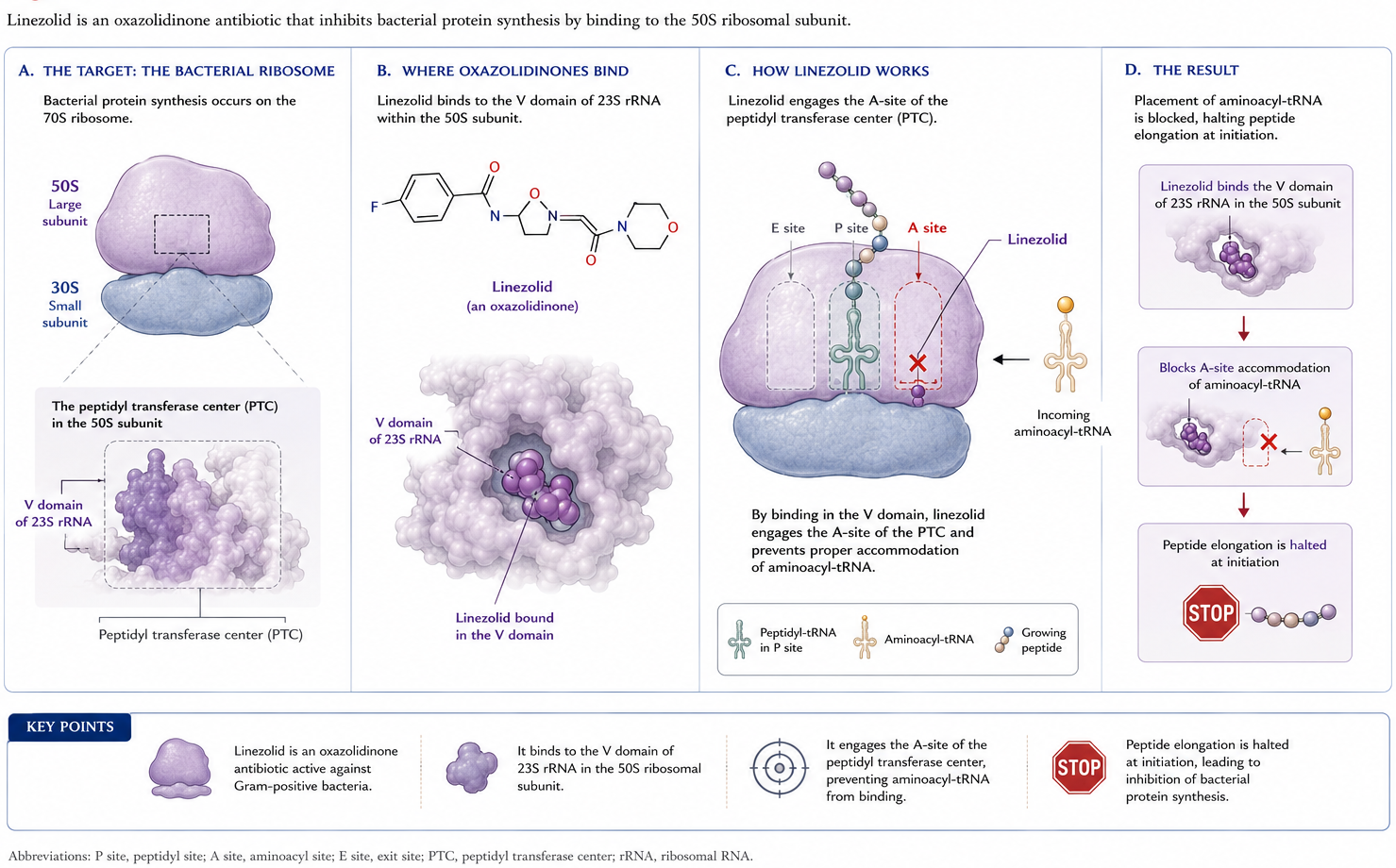
The oxazolidinones are inhibitors of protein synthesis and are usually bacteriostatic, although in some models bactericidal activity against select organisms has been observed. The mechanism is unique: oxazolidinones inhibit the earliest steps of bacterial protein synthesis (106). They bind the V domain of the 23S rRNA component of the 50S ribosomal subunit, and interaction with the A-site of the peptidyl transferase center blocks peptide elongation (Figure 2) (37,126). Binding is competitively inhibited by chloramphenicol and lincomycin owing to proximity of the respective binding sites (106).
Figure 2: Mechanism of action of oxazolidinones. Oxazolidinones bind the V domain of the 23S rRNA of the 50S ribosomal subunit, blocking peptide elongation at the peptidyl transferase center (PTC). Modified from (37).
5 Antimicrobial Activity
5.1 General considerations
In vitro studies have established MICs of linezolid and tedizolid against a wide variety of organisms. In general, comparisons show tedizolid to be two- to eight-fold more potent(27,51,85,126,169). Lower MICs may not translate into superior clinical activity, however; only direct clinical comparisons can establish that.
5.2 Activity against Gram-positive organisms
Both linezolid and tedizolid demonstrate consistent activity against most clinically important Gram-positive organisms: S. aureus (MSSA, MRSA, vancomycin-intermediate, vancomycin-resistant); coagulase-negative staphylococci; E. faecalis and E. faecium (vancomycin-susceptible and vancomycin-resistant); and streptococci including penicillin-resistant S. pneumoniae (Table 1) (29,30,36,37,51,58,86,126). Tedizolid consistently has lower MICs across these organisms.
CLSI has established breakpoints for select Gram-positive cocci, noting that tedizolid susceptibility can be inferred for S. aureus, E. faecalis, S. agalactiae, S. pyogenes, and S. anginosus isolates that test susceptible to linezolid. Linezolid-resistant strains may still be tedizolid-susceptible (36). EUCAST offers similar guidance for staphylococci and β-hemolytic streptococci (54).
A variety of other Gram-positive organisms may be susceptible to linezolid or tedizolid, though fewer isolates have been evaluated: Corynebacterium spp., Listeria monocytogenes, Bacillus spp., Micrococcus spp., Erysipelothrix rhusiopathiae, Leuconostoc spp., Rhodococcus equi, and Pediococcus spp. (3,51).
Table 1: In vitro susceptibility of common aerobic Gram-positive organisms to linezolid and tedizolid. Compiled from (29,30,36). CLSI susceptibility breakpoints: staphylococci ≤4 μg/mL (linezolid), ≤0.5 μg/mL (tedizolid, S. aureus only); β-hemolytic streptococci ≤2 μg/mL (linezolid), ≤0.5 μg/mL (tedizolid, S. pyogenes and S. agalactiae only); viridans streptococci ≤2 μg/mL (linezolid), ≤0.25 μg/mL (tedizolid); enterococci ≤2 μg/mL (linezolid), ≤0.5 μg/mL (tedizolid, E. faecalis only). Dashes indicate no determined breakpoint.
Organism
Linezolid MIC₉₀ (μg/mL)
Linezolid % susceptible
Tedizolid MIC₉₀ (μg/mL)
Tedizolid % susceptible
S. aureus — oxacillin-susceptible
2
100
0.25
100
S. aureus — oxacillin-resistant
2
>99.9
0.25
>99.9
S. agalactiae
2
100
0.25
100
S. anginosus group
1
100
0.25
100
S. pyogenes
2
100
0.25
100
E. faecalis
2
99.5
0.25
99.9
E. faecalis — linezolid-nonsusceptible
8
0
1
73.1
E. faecium
2
99.1
0.5
—
5.3 Activity against higher-order bacteria
Linezolid has in vitro activity against a wide range of Nocardia spp., with MIC₉₀ values of 1–4 μg/mL (82). Virtually all strains tested are susceptible (67,165). Tedizolid demonstrates MIC₉₀ values several-fold lower than linezolid for many species, with the exception of Nocardia nova complex and Nocardia brasiliensis, where values are comparable (25). Limited data suggest that some actinomycetes may also be susceptible (25).
5.4 Activity against Mycobacterium spp.
Oxazolidinones have demonstrated activity against M. tuberculosis, including multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains. Linezolid MIC range is approximately 0.125–2 μg/mL, even for MDR and XDR isolates (4,5,106,162). Tedizolid MICs typically range from 0.125–0.5 μg/mL (4,125). Resistance to linezolid in MDR-TB has been reported but appears uncommon (142).
Both agents also have in vitro activity against nontuberculous mycobacteria. MICs for rapid growers such as M. abscessus subsp. abscessus and M. abscessus subsp. massiliense and for the slow grower M. kansasii are lower than for M. avium and M. intracellulare(5,24,26,79,106,149). Tedizolid appears more potent for the strains tested, but the clinical significance is unclear (26).
5.5 Activity against other organisms
The oxazolidinones have limited activity against aerobic Gram-negatives. Assessment of respiratory pathogens shows that both agents provide incomplete coverage for Haemophilus influenzae and Moraxella catarrhalis(15,106,169). Both oxazolidinones possess activity against Gram-positive and Gram-negative anaerobes, including Clostridium spp. and Bacteroides spp. (30,51,65).
WarningPractical implication
Because oxazolidinones lack reliable Gram-negative activity, do not use linezolid or tedizolid as monotherapy for empirical nosocomial pneumonia or intra-abdominal infection — pair with an agent that covers Gram-negatives.
6 Resistance
Resistance to linezolid was reported even before the drug was released in the United States, among patients treated for vancomycin-resistant enterococci (VRE) under the Linezolid Compassionate Use Program (16). Despite use in the United States since 2000, resistance remains <1% of clinical isolates (101). Enterococci and, to a lesser extent, staphylococci are the organisms most often associated with resistance; prior drug exposure and prolonged therapy are the most common predisposing factors (8,61,100). Although unusual, outbreaks due to linezolid-resistant bacteria are well described (19,43,112,117,130).
Similar susceptibility to tedizolid has been observed over a 5-year surveillance period, with activity against some linezolid-resistant strains. Tedizolid resistance is less frequent than linezolid resistance (29,30).
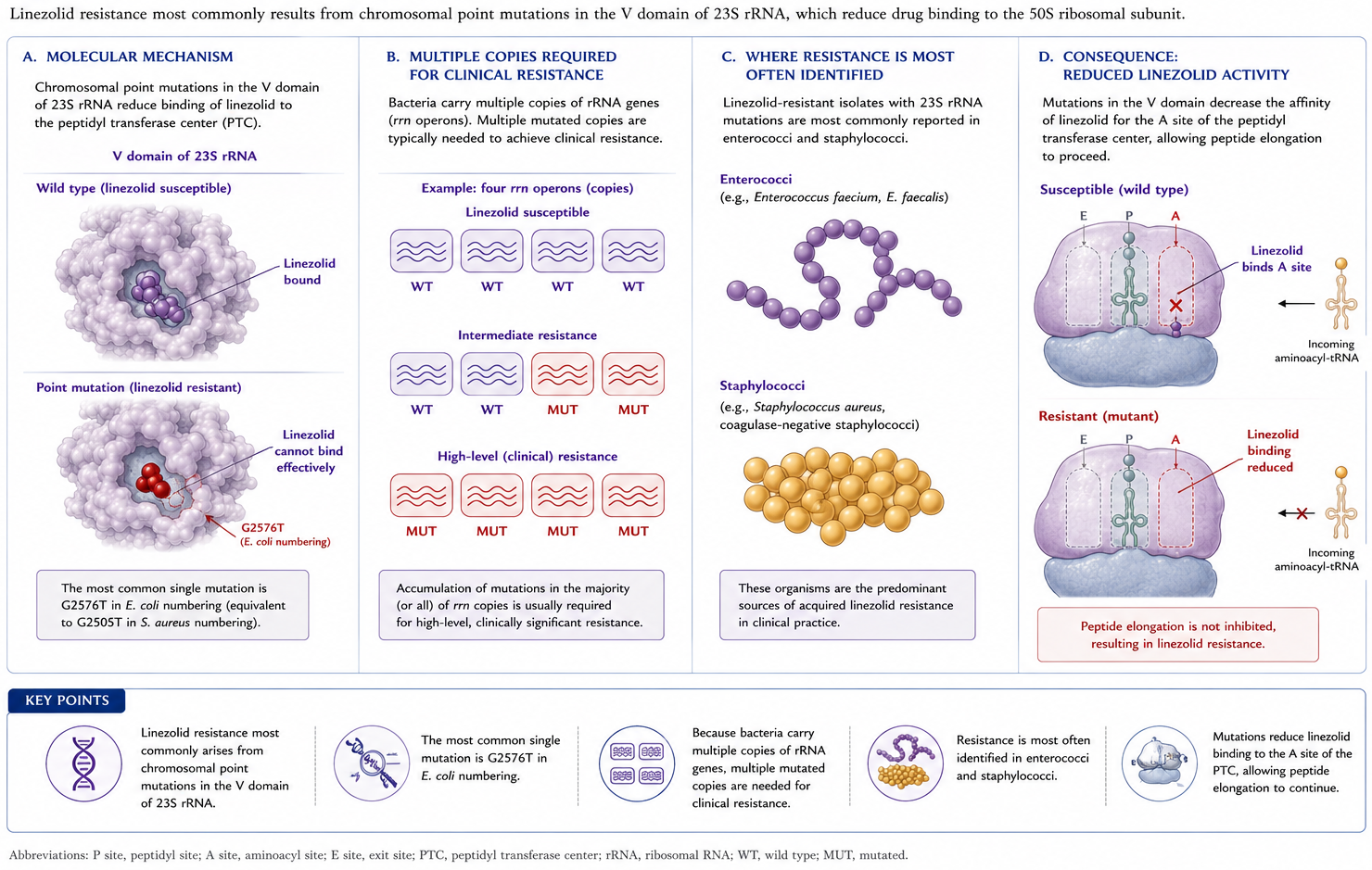
6.1 Mechanism 1 — 23S rRNA mutations
A frequent mechanism involves chromosomal mutations of the oxazolidinone binding site on the V region of 23S rRNA (Figure 3). Because bacteria carry numerous copies of the gene, multiple mutations are needed for clinical resistance. This is most often identified in enterococci and staphylococci (87).
Figure 3: Mechanism 1: Chromosomal mutations in the V region of 23S rRNA reduce oxazolidinone binding affinity. Because bacteria carry multiple gene copies, several simultaneous mutations are required for clinical resistance.
6.2 Mechanism 2 — ribosomal protein mutations
Additional binding-site modifications come from mutations in genes encoding the 50S ribosomal proteins L3, L4, and possibly L22 (Figure 4). These have been implicated in resistance in staphylococci, S. pneumoniae, and Clostridium perfringens, among others (21,68,87).
Figure 4: Mechanism 2: Mutations in ribosomal proteins L3, L4, and L22 alter the geometry of the oxazolidinone binding pocket.
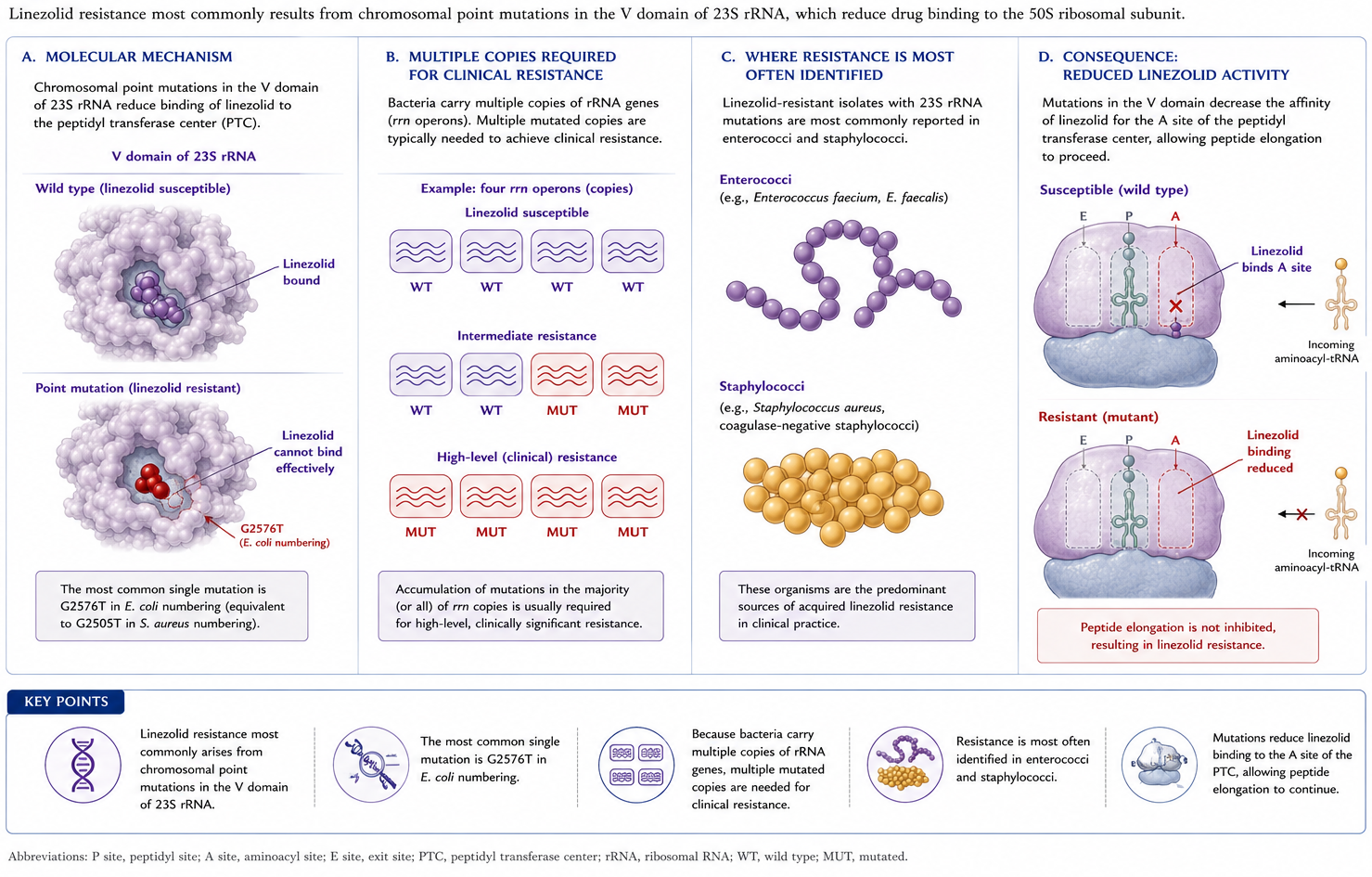
6.3 Mechanism 3 — cfr methyltransferase
Acquisition of mobile genetic elements is another important mechanism. The cfr (chloramphenicol-florfenicol resistance) gene encodes a ribosomal RNA methyltransferase that confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibacterial agents (the PhLOPSₐ phenotype; Figure 5) (50). It has been identified in a large number of Gram-positive organisms (enterococci, staphylococci, Bacillus spp.) and some Gram-negatives; cfr-like genes confer the same phenotype (21).
Figure 5: Mechanism 3: The cfr methyltransferase methylates position A2503 of 23S rRNA, conferring the PhLOPSₐ multi-drug resistance phenotype (Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, Streptogramin A).
ImportantWhy tedizolid’s structure matters here
Tedizolid’s hydroxymethyl C5 side chain allows it to retain activity against many cfr-positive isolates of S. aureus, coagulase-negative staphylococci, and enterococci. However, tedizolid resistance may emerge when cfr is combined with chromosomal ribosomal mutations (27).
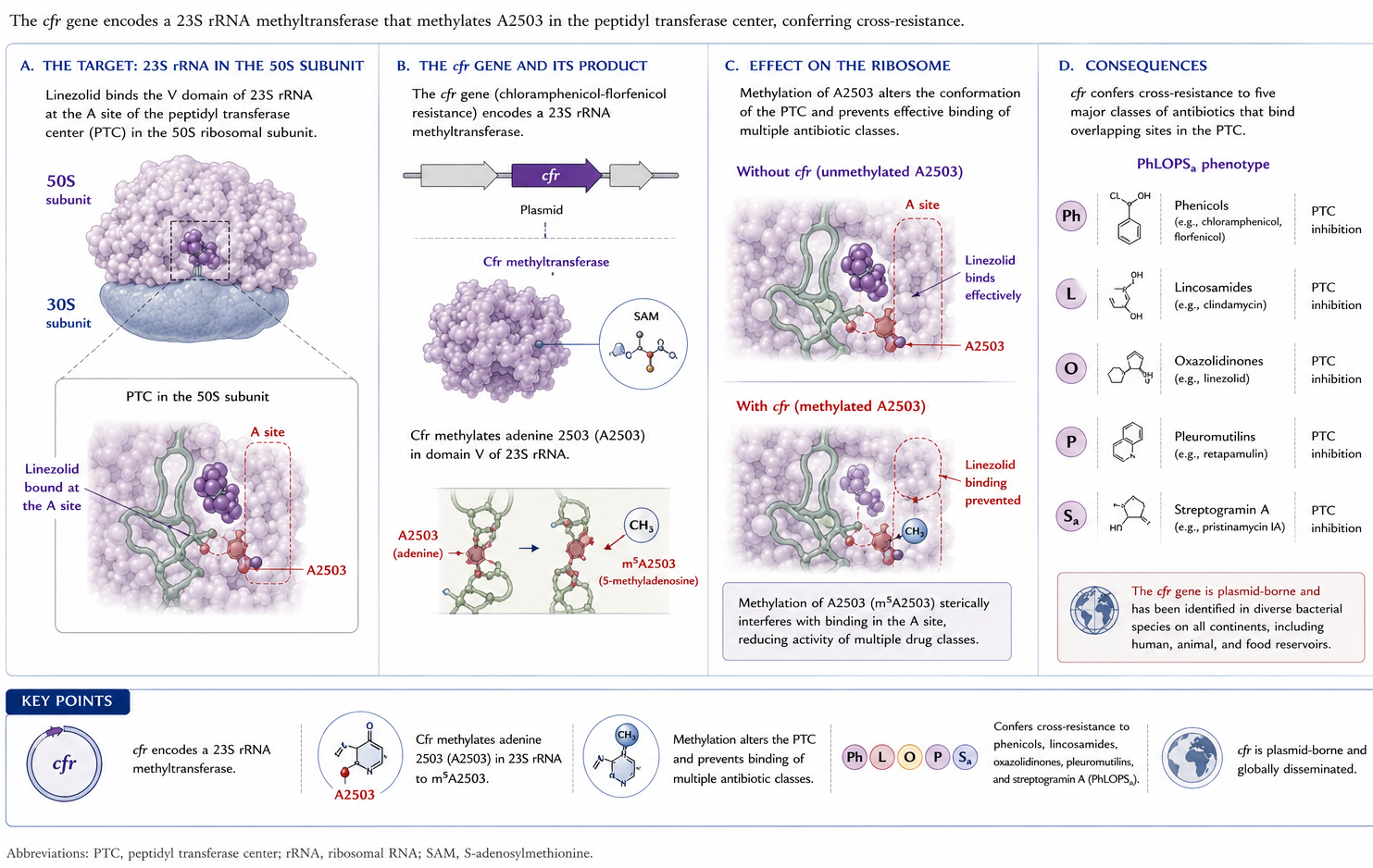
6.4 Mechanism 4 — optrA and poxtA
Other plasmid-borne resistance genes include optrA and poxtA, which encode an ABC-F protein that mediates resistance through target protection (Figure 6) (13,21). optrA has been identified most commonly in enterococci (including a widespread presence among isolates of animal origin), streptococci, and staphylococci, whereas poxtA has been found primarily in enterococci (13,21,151).
Figure 6: Mechanism 4: optrA and poxtA encode ABC-F ribosomal protection proteins that displace oxazolidinones from the ribosomal binding site.
6.5 Other resistance mechanisms
Other mechanisms include active efflux, biofilm production, and loss of activity of the rlmN gene, which encodes an RNA methyltransferase (21).
7 Pharmacology
7.1 Linezolid
The approved adult/adolescent dose of linezolid is 600 mg intravenously or orally every 12 hours for serious infections, with 400 mg every 12 hours for adults with uncomplicated SSTI. Pediatric dosing is generally 10 mg/kg every 8 hours in patients from birth to 11 years owing to increased weight-based clearance; the frequency is reduced to every 12 hours in children 5–11 years for uncomplicated SSTI.
Absorption after ingestion is rapid, with peak serum levels at 1–2 hours and bioavailability approaching 100%. The standard 600 mg twice-daily regimen yields peak serum concentrations of 15.1 μg/mL (oral) and 21.2 μg/mL (IV) — well above the MIC₉₀ for usual targets (116).
Distribution. Linezolid is 31% bound to plasma proteins. It distributes readily into well-perfused tissues, achieving adequate concentrations in pulmonary epithelial lining fluid, alveolar macrophages, and skin and soft tissue(116,124). Trough CSF concentrations in post-neurosurgical patients and in one patient with meningitis ranged from <0.2 to 7.0 μg/mL with CSF/plasma ratios of 0.77–1.0(88,146,168). Peak CSF concentrations of 3.12–12.5 μg/mL have been documented in meningitis (131,168). Bone and joint tissue penetration is variable (80,124).
Metabolism and clearance. Linezolid is metabolized by oxidation and interacts minimally with the common drug-metabolizing CYP450 enzymes (114). Approximately 65% of total clearance is nonrenal; 30% is excreted unchanged in urine, and fecal excretion of two inactive carboxylic acid metabolites accounts for most of the remainder (116,124). Elimination half-life in adults is 5–7 hours(124). No dose adjustment is suggested in product labeling for renal or mild-to-moderate hepatic impairment, but because linezolid and its metabolites are removed by dialysis, administration after hemodialysis is suggested (116). Continuous renal replacement therapy also removes drug; no routine dose change is definitively recommended (11,49).
Limited case-report data have not signaled adverse outcomes in pregnancy; however, given fetal harm in animals, linezolid in pregnancy should be used only when the potential benefit outweighs the potential fetal risk(116).
TipPK/PD targets
The parameters most predictive of efficacy are the time above MIC (T > MIC ≥ 85%) and the AUC/MIC ratio (target 80–120)(124). Large interindividual variability with standard dosing may threaten target attainment (32).
Pharmacokinetic changes in special populations may predispose to:
Recommendations for therapeutic drug monitoring (TDM) have therefore gained momentum (2). A target steady-state trough concentration (Cmin) of 2–7 mg/L has been proposed for Gram-positive infection, whereas a Cmin<2 μg/mL has been proposed for TB infection (where MICs are typically lower) (11). Further study is needed to standardize TDM targets and dose-adjustment algorithms.
Drug interactions. Although mostly free of CYP interactions, linezolid does interact with several agents:
Rifampin → 32% decrease in linezolid AUC in healthy volunteers (60)
These effects may reflect linezolid acting as a P-glycoprotein substrate(18,115). Interactions with serotonergic and adrenergic agents are discussed under Untoward Reactions.
7.2 Tedizolid
Tedizolid phosphate is a prodrug converted in vivo by plasma phosphatases to active tedizolid after oral or IV administration. The phosphate group enhances aqueous solubility, and an absolute bioavailability of 91% permits no dosage adjustment between formulations. The approved dose for adults and pediatric patients ≥ 12 years is 200 mg orally or intravenously once daily. Peak serum concentrations following 200 mg oral and IV doses are 1.8–2.4 μg/mL and 2.6–3.5 μg/mL, respectively. The half-life of ~12 hours supports once-daily dosing, and the oral product can be taken without regard to meals.
Distribution. Plasma protein binding is 70–90%, whereas adipose and skeletal muscle penetration yields exposures similar to free plasma drug. Relative pulmonary distribution in healthy volunteers is high, with AUC ratios of epithelial lining fluid and alveolar macrophage to plasma of 40 and 20, respectively.
Metabolism and excretion. Tedizolid is primarily hepatically metabolized and eliminated in feces as an inactive sulfate conjugate; <3% is excreted unchanged in urine. Pharmacokinetic parameters are similar for patients with hepatic impairment, renal insufficiency (including hemodialysis), and obesity (BMI ≥ 30 kg/m²)(71,103,169). No dose adjustment is required in these special populations. Pregnant patients should be advised of potential risk given that fetal harm was identified in animal studies (103).
WarningTedizolid and neutropenia
Free AUC/MIC ratio most closely predicts tedizolid efficacy in murine models, but antistaphylococcal activity is markedly decreased in granulocytopenic mice(53). Higher exposure is required for equivalent effect in pneumococcal lung models in the absence of granulocytes, although less pronounced (1). Clinically relevant doses against S. aureus in neutropenic mice achieve stasis, but the product label retains a precaution to consider alternative therapies in neutropenic patients given limited human data (72,103,161).
Drug interactions. Tedizolid does not appreciably interact with CYP450 enzymes but inhibits intestinal breast cancer resistance protein (BCRP) and increases serum concentrations of orally administered BCRP substrates (e.g., rosuvastatin, methotrexate). Monitor for BCRP-substrate–related adverse reactions if coadministration cannot be avoided (103). Serotonergic and adrenergic drug-interaction potential is discussed under Untoward Reactions.
8 Clinical Use
8.1 Linezolid — FDA-approved indications
Linezolid was approved by the FDA in April 2000 for adults and children with a variety of clinical infections involving susceptible Gram-positive organisms (116):
Nosocomial pneumonia caused by S. aureus (MRSA or MSSA) or S. pneumoniae
Community-acquired pneumonia caused by S. pneumoniae, including cases with concurrent bacteremia or MSSA
Complicated skin and skin structure infections, including diabetic foot infections without concomitant osteomyelitis caused by S. aureus (MRSA or MSSA), S. pyogenes, or S. agalactiae
Uncomplicated skin and skin structure infections caused by MSSA or S. pyogenes
Vancomycin-resistant E. faecium infections, including those with concurrent bacteremia
8.2 Tedizolid — FDA-approved indications
Tedizolid was FDA-approved in 2014 for adults and pediatric patients ≥ 12 years for treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible Gram-positive organisms, including MRSA and MSSA, S. pyogenes, S. agalactiae, S. anginosus group, and E. faecalis(103).
8.3Staphylococcus aureus including MRSA
8.3.1 Linezolid
Although levels of evidence vary, guidelines recommend linezolid as initial or alternative therapy for complicated SSTI, pneumonia, bone and joint infections, and CNS infections (23,84).
SSTI. Linezolid is an important option for SSTI, particularly MRSA. A Cochrane review of 9 RCTs comparing linezolid versus vancomycin for SSTI found superior clinical and microbiologic cure rates, shorter hospital stays, and lower overall cost with linezolid. Subgroup analysis revealed superiority only in adults, not in patients <18 years; methodologic quality was poor with potential for publication bias (167). A subsequent network meta-analysis of RCTs (linezolid vs vancomycin, daptomycin vs vancomycin, tedizolid vs linezolid) found linezolid clinically and microbiologically superior to vancomycin, with no significant differences between other agents and no significant difference in adverse events between agents in this analysis (55).
Nosocomial pneumonia. Pooled data from two RCTs suggested that linezolid plus aztreonam was equivalent to vancomycin plus aztreonam for nosocomial pneumonia, with subgroup analysis suggesting higher cure rates and improved survival in MRSA infection (159). The subgroup analysis — and therefore the conclusion — has been questioned by the FDA(119).
A subsequent prospective, double-blind, multicenter study by the same group (ZEPHyR) showed higher clinical success with linezolid but no difference in 60-day mortality. Interpretation was complicated by more patients in the vancomycin arm being mechanically ventilated and having MRSA bacteremia, and by suboptimal vancomycin trough concentrations (158). In a subsequent meta-analysis of nine RCTs, including one specifically evaluating MRSA nosocomial pneumonia, no differences in clinical or microbiologic efficacy or all-cause mortality were noted. The only significant toxicity difference was higher nephrotoxicity with vancomycin(77,152). Although none of the studies is definitive, this analysis appears the most complete at present.
Bacteremia. Linezolid is effective for both MSSA and MRSA bacteremia in selected patients, including those with persistent bacteremia, vancomycin failure, or both (75,81,84). Clinical hesitancy because of linezolid’s bacteriostatic rather than bactericidal effect has not been supported by careful review (148). Oral step-down from parenteral to oral linezolid appears as effective as exclusive parenteral therapy in staphylococcal bacteremia, including MRSA (44,157,164). Which patients would benefit is unsettled — most studies have involved uncomplicated bacteremia.
Endocarditis. Use is evolving. In the POET trial, linezolid was used as part of a two-drug oral step-down regimen for stable left-sided IE after at least 10 days of parenteral therapy; partial oral therapy was noninferior to parenteral therapy (73). MRSA endocarditis was not evaluated. Other studies have shown inconsistent results with linezolid for endovascular infections with MRSA, including endocarditis, some showing increased mortality (109). Guidelines do not list linezolid as suggested therapy for MRSA endocarditis(7,153).
8.3.2 Tedizolid
Major clinical use of tedizolid thus far has been in SSTI. Two trials (ESTABLISH-1 and ESTABLISH-2) compared tedizolid 200 mg once daily × 6 days with linezolid 600 mg twice daily × 10 days; S. aureus was the most common pathogen, with MRSA in 27–42%. Tedizolid was noninferior to linezolid at 48–72 hours and at end of therapy. Drug-related toxicity was overall similar, but GI adverse effects and thrombocytopenia were less common with tedizolid (107,120). Pooled analysis of both studies reached the same conclusions (135). Subsequent trials have reported comparable efficacy (90,104); GI and hematologic effects were more common with linezolid, while thrombophlebitis was more common with tedizolid.
Tedizolid has also been compared with other ABSSSI agents. In a network meta-analysis of 15 RCTs, the clinical response to tedizolid was superior to vancomycin but comparable to linezolid, tigecycline, ceftaroline, teicoplanin, daptomycin, and telavancin (96). A Phase III RCT in an adolescent population showed similar efficacy and safety (20).
A Phase III RCT of tedizolid versus linezolid for adults with Gram-positive hospital-acquired or ventilator-associated pneumonia showed that tedizolid was noninferior to linezolid for the primary endpoint of day-28 all-cause mortality; however, noninferiority for investigator-assessed clinical response was not reached — the reason was unclear. Drug-related side effects were similar (160).
8.4 Coagulase-negative staphylococci
8.4.1 Linezolid
Case reports and case series describe cure after linezolid for bone and joint infection, meningitis, ventriculoperitoneal shunt infections, and endocarditis caused by coagulase-negative staphylococci, often in the setting of prosthetic material (92,113,122,138). There are inadequate data to recommend linezolid routinely as initial treatment. A small number of CoNS endocarditis cases were treated successfully with oral two-drug step-down therapy in POET, after ≥10 days of parenteral therapy (73).
8.4.2 Tedizolid
Although small numbers of CoNS infections have been treated with tedizolid, no statements can be made regarding its overall clinical utility.
8.5 Vancomycin-resistant enterococci
8.5.1 Linezolid
Linezolid has been shown to be effective for VRE infection. Successful treatment has been reported for:
Linezolid has been compared with daptomycin for VRE bacteremia in several meta-analyses initially suggesting linezolid’s superiority; inclusion of patients treated with low daptomycin doses limited interpretation (10,35,97). A more recent meta-analysis including 22 observational studies showed a nonsignificant increase in mortality with daptomycin, with no significant difference in clinical response, microbiologic cure, or recurrence; subgroup analysis using only daptomycin doses >6 mg/kg showed no significant mortality difference (133). A retrospective US Department of Veterans Affairs cohort study found contradictory results favoring daptomycin — higher clinical success and lower 30-day mortality — even at relatively low doses (22).
NoteBottom line for VRE bacteremia
There is no clear “best” therapy. Antibiotic selection should be individualized based on in vitro susceptibility, antecedent antibiotic exposure, source of infection, comorbidities, and potential for drug-related toxicity.
For VRE endocarditis, both treatment success and failure have been described. Linezolid (and daptomycin) is recommended for endocarditis caused by enterococci resistant to penicillin, aminoglycosides, and vancomycin — usually E. faecium — but the strength of the recommendation is limited by small patient numbers (7). Animal data suggest possible synergy of linezolid with gentamicin, doxycycline, ceftriaxone, and daptomycin; clinical combination therapy with linezolid for VRE infection is unproven(33).
8.5.2 Tedizolid
In vitro and pharmacodynamic properties suggest tedizolid would be useful for VRE infections, including linezolid-resistant strains. Only case reports and abstracts are available to date; overall efficacy is yet to be determined (136,141).
8.6 Streptococci including S. pneumoniae
8.6.1 Linezolid
Efficacy against S. pneumoniae has been demonstrated in randomized open-label trials of community-acquired pneumonia in hospitalized adults and children (78,128). Despite approval for pneumococcal CAP, limited activity against H. influenzae and inconsistent activity against M. pneumoniae and Chlamydia pneumoniae should preclude linezolid as first-line empirical CAP therapy(106).
Data describing linezolid efficacy for pneumococcal CNS infection are limited to case reports/series, largely with linezolid combined with ceftriaxone for penicillin-nonsusceptible isolates. In one case a postsurgical cerebral abscess caused by penicillin-susceptible S. pneumoniae was treated successfully with linezolid (113). Linezolid has also been used successfully for streptococcal SSTI including S. pyogenes and S. agalactiae(3).
TipLinezolid as a clindamycin substitute in group A streptococcal toxic shock?
The role of linezolid as a substitute for clindamycin as adjunctive therapy in group A streptococcal necrotizing soft tissue infection and toxic shock has been reviewed (41). Clindamycin’s role is itself debated but is recommended in necrotizing-fasciitis guidelines (140) and supported by recent literature (6). Like clindamycin, linezolid inhibits protein synthesis and suppresses streptococcal exotoxin release. Clinical experience with linezolid in this role is limited; use has been suggested when clindamycin resistance is encountered.
8.6.2 Tedizolid
Streptococcal SSTI has been successfully treated with tedizolid in the ESTABLISH-1 and ESTABLISH-2 trials; S. pyogenes, S. anginosus group organisms, S. agalactiae, and Gemella morbillorum were isolated alongside S. aureus(107,120). Response rates were lower with tedizolid in S. anginosus group infections compared with linezolid (70% vs 89%) — although numbers were insufficient to determine statistical significance (135). The role of tedizolid for other streptococcal infections, including S. pneumoniae, has not been established.
8.7Mycobacterium spp. including M. tuberculosis
8.7.1 Linezolid
Linezolid as therapy for mycobacterial infection represents one of its most important newly defined roles. It demonstrates bacteriostatic activity against M. tuberculosis, including MDR and XDR strains. In vitro studies with established first- and second-line agents usually reveal additive rather than synergistic interactions (91,170).
In combination with the newer oral agents bedaquiline and pretomanid, linezolid at 1200 mg/day has produced favorable outcomes in approximately 90% of patients with resistant tuberculosis (Nix-TB and related cohorts) (39,69). The WHO has suggested this regimen could be used for 6–9 months for treatment of resistant TB (105). Toxicity at this dose, however, occurred in >80% of patients — peripheral neuropathy and myelosuppression were most common. Close monitoring and dose reduction were recommended to mitigate adverse events, most of which were reversible (70).
A subsequent randomized study (ZeNix) showed that reducing the linezolid dose to 600 mg/day for 26 weeks led to fewer toxicity episodes and less need for dose modification, while maintaining favorable outcomes at 91% — only slightly lower than the 94% response with the 1200 mg/day dose (38).
ImportantModern BPaL dosing
For drug-resistant TB, the 600 mg/day × 26 week linezolid dose within the BPaL regimen offers the best balance of efficacy and tolerability (38,70).
Linezolid has been used successfully in nontuberculous mycobacterial infections in immunocompromised patients, although large-scale studies are lacking (98,118).
8.7.2 Tedizolid
In vitro models suggest tedizolid could be effective for M. tuberculosis and NTM, but few clinical data are available. A single series showed that tedizolid was comparable to linezolid as part of combination therapy for NTM infections in solid-organ transplant recipients (118).
8.8Nocardia spp.
8.8.1 Linezolid
Based on near-uniform activity against all species, bioavailability, and CNS penetration, linezolid has been used successfully in a variety of nocardiosis cases. It has been suggested as an alternate regimen combined with imipenem for pulmonary, disseminated, and CNS infection, and as monotherapy for primary skin disease (93). Linezolid has been used as combination induction therapy (usually with TMP-SMX and ceftriaxone) for disseminated, cutaneous, or moderate-severe pulmonary disease, with 1-year survival of approximately 85% — comparable to other regimens (45). The major linezolid-related toxicity was thrombocytopenia. Linezolid has been similarly used for CNS nocardiosis (40). Use in organ transplant and hematopoietic stem cell transplant recipients has been described in small series (42,47). Although usually combined, monotherapy has been suggested to avoid cumulative myelotoxicity with TMP-SMX and potential antagonism with amikacin (93).
8.8.2 Tedizolid
Few data exist on tedizolid for Nocardia infections. In case reports and small series, it has been used successfully in CNS, pulmonary, and disseminated disease (34,94,139). Comparative efficacy and safety relative to linezolid require further testing.
9 Oral Versus Intravenous Administration
Because bioavailability of both agents approaches 100%, oral preparations may substitute for IV therapy. When oral dosing can safely be used, the decision should depend on:
patient stability and reliability
ability to absorb oral medications
clinical experience reported in peer-reviewed literature
10 Untoward Reactions
Both linezolid and tedizolid are relatively well tolerated, with headache, diarrhea, nausea, and vomiting predominating in larger clinical trials (103,116). Serious but less common adverse events are discussed below.
10.1 Hematologic toxicity
Reversible myelosuppression — including pure red cell aplasia, pancytopenia, and especially thrombocytopenia — has been clearly documented with linezolid (64). Thrombocytopenia is the most common manifestation; although the mechanism is not definitively established, both a drug-induced immune-mediated phenomenon and inhibition of platelet progenitor cells have been suggested (14,17). Adults completing Phase III clinical trials developed thrombocytopenia in 2.4% of cases (range 0.3–10%), but risk is enhanced by:
Delayed neutrophil recovery and worsening thrombocytopenia have been documented in patients with baseline marrow suppression (66,74). Anemia appears to be caused by suppression of normal erythropoiesis, similar to the marrow effect of chloramphenicol (66).
WarningMonitoring
Weekly CBC monitoring is recommended, particularly for therapeutic durations exceeding 2 weeks (116). Therapeutic drug monitoring may provide additional context — hematologic toxicity has been associated with trough concentrations above 7.5–22.1 mg/L(11,121).
Thrombocytopenia occurred in 2.1% of patients treated with tedizolid for 6 days, compared with 3.8% receiving linezolid for 10 days in pooled Phase III ABSSSI analysis (103). Among 81 patients prescribed tedizolid for a median of 28 days (mostly to avoid linezolid toxicity or interactions), 6 developed thrombocytopenia after a median 26.5 days; only 2 discontinued therapy (102). In more recent evaluations, no attributable hematologic toxicity occurred among small numbers receiving tedizolid for a median of 6 months. One center reported reversible myelosuppression for all 8 patients with linezolid-associated myelotoxicity at baseline after transition to tedizolid (56,108).
10.2 Monoamine oxidase inhibition
Linezolid is a reversible, nonselective monoamine oxidase inhibitor and has been associated with serotonin syndrome (fever, agitation, mental status changes, tremors) in patients receiving concurrent serotonergic agents (111,116). In an FAERS analysis of drug-interaction–associated serotonin syndrome events, citalopram, escitalopram, and methadone were implicated as highest risk (63). Overall, serotonin syndrome associated with linezolid drug interactions appears rare, yet risks and benefits should be carefully weighed for each individual patient (143).
Small increases in systolic blood pressure have been documented with concurrent tyramine, leading to dietary-restriction recommendations. Blood pressure monitoring is similarly recommended for hypertensive patients receiving linezolid or during coadministration of adrenergic agents(116).
Tedizolid exhibits weak, reversible monoamine oxidase inhibition in vitro. Although patients on concurrent serotonergic agents were excluded from Phase II and III studies, neither human nor animal investigation revealed adverse hypertensive or serotonergic effects at therapeutic dosing (124). Four cases of tedizolid-associated serotonin syndrome have been reported to FAERS without further characterization (62).
10.3 Adverse effects associated with mitochondrial toxicity
Disruption of mitochondrial protein synthesis is the proposed explanation for linezolid-induced neuropathies and lactic acidosis, both detailed below (12,48,111). Case reports of suspected linezolid-associated rhabdomyolysis and drug-induced liver injury have also raised the possibility of mitochondrial etiology (28,46).
Although tedizolid is thought to be a more potent mitochondrial protein synthesis inhibitor, less free drug exposure and a recovery window during each dosing interval may reduce risk (57). More study is needed to confirm these findings in humans.
10.4 Neuropathy
Linezolid-induced neuropathies are well documented, particularly during prolonged therapy(38,39,111):
Peripheral neuropathy may begin with dysesthesias in the hands and is often poorly reversible
Optic neuropathy causes gradual onset of blurring and can lead to permanent loss of useful visual acuity if the drug is not discontinued; when detected early, visual loss has generally been reversible
Whether TDM will ultimately assist in managing patients at highest risk of neuropathies remains unclear (70,76). Peripheral neuropathy and optic nerve disorders in Phase III studies of patients receiving tedizolid (6 days) or linezolid (10 days) occurred with similar frequency (103). Where long-term tedizolid use has been described, reports of neuropathy have been absent or — when present — not definitively drug-related (56,62,108,132). Additional safety data are needed.
10.5 Lactic acidosis
Lactic acidosis — including fatal cases — has been reported most commonly during prolonged linezolid therapy but can develop within the first week(111,129,145). Prompt recognition and drug discontinuation are critical. Age, renal insufficiency, and drug interactions causing linezolid overexposure increase risk. A genetic predisposition has also been suggested (129). Lactic acidosis during tedizolid treatment has been reported to FAERS, but a direct association is less clear (62).
10.6 Miscellaneous untoward reactions
In patients treated with linezolid for intravascular catheter-related infections, increased mortality was observed in the setting of Gram-negative infection, mixed Gram-positive/Gram-negative infections, or no clear infection at the time of therapy (156). The poor Gram-negative activity of linezolid should limit its use as a single agent in such situations; increased mortality with no clear infection remains unexplained.
Other notable but rare post-marketing adverse effects with linezolid include:
tooth and tongue discoloration; black hairy tongue (lingua villosa nigra) (123)
hypoglycemia among diabetic patients receiving insulin or oral hypoglycemic agents (116,147)
Finally, linezolid oral suspension contains phenylalanine and could be harmful to patients with phenylketonuria (116).
11 Other Oxazolidinones
The ability to modify the basic oxazolidinone structure has led to the development of a series of compounds presently being evaluated (58). Although in vitro activity has been studied, clinical efficacy and safety investigations are ongoing. Agents of potential interest:
Radezolid — increased activity against Gram-positives compared with linezolid, including linezolid-resistant strains (83)
Sutezolid — structurally similar to linezolid, with potent activity against mycobacteria
Delpazolid — activity against Gram-positives similar to linezolid, with superior in vitro activity against mycobacteria(155,166)
Contezolid (MRX-I) — activity similar to linezolid against a variety of Gram-positives including MRSA, VRE, and Mycobacterium spp. (134,150)
TipTake-home points
Oxazolidinones are a synthetic class binding the 23S rRNA / PTC of the 50S ribosome, with no cross-resistance with β-lactams, glycopeptides, or daptomycin
Two FDA-approved agents: linezolid (Zyvox, BID, ~100% oral bioavailability) and tedizolid (Sivextro, once-daily prodrug, retains activity against many cfr-positive strains)
Resistance is rare but rising — chromosomal 23S/L3-L4-L22 mutations, cfr, optrA/poxtA; risk factors are prior exposure and prolonged therapy
TDM with target trough 2–7 mg/L is helpful in critically ill patients, renal failure, hepatic dysfunction, and prolonged therapy
For drug-resistant TB, linezolid 600 mg/day × 26 weeks within BPaL balances efficacy and tolerability
References
1.
Abdelraouf K, Nicolau DP. Comparative in vivo efficacies of tedizolid in neutropenic versus immunocompetent murine Streptococcus pneumoniae lung infection models. Antimicrobial Agents and Chemotherapy. 2016;27:e01957–16. doi:10.1128/AAC.01957-16
2.
Abdul-Aziz MH, Alffenaar JC, Bassetti M, et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A position paper. Intensive Care Medicine. 2020;46(6):1127–53. doi:10.1007/s00134-020-06050-1
3.
Ager A, Gould K. Clinical update on linezolid in the treatment of gram-positive bacterial infections. Infection and Drug Resistance. 2012;5:87–102. doi:10.2147/IDR.S25890
4.
Aono A, Murase Y, Chikamatsu K, et al. In vitro activity of tedizolid and linezolid against multidrug-resistant Mycobacterium tuberculosis: A comparative study using microdilution broth assay and genomics. Diagnostic Microbiology and Infectious Disease. 2022;103(3):115714. doi:10.1016/j.diagmicrobio.2022.115714
5.
Ashtekar DR, Costa-Periera R, Shrinivasan T, et al. Oxazolidinones, a new class of synthetic antituberculosis agent: In vitro and in vivo activities of DuP-721 against Mycobacterium tuberculosis. Diagnostic Microbiology and Infectious Disease. 1991;14(6):465–71. doi:10.1016/0732-8893(91)90002-w
6.
Babiker A, Li X, Ling Lai Y, et al. Effectiveness of adjunctive clindamycin in \(\beta\)-lactam antibiotic-treated patients with invasive \(\beta\)-haemolytic streptococcal infections in US hospitals: A retrospective multicenter cohort study. The Lancet Infectious Diseases. 2021;21(5):697–710. doi:10.1016/S1473-3099(20)30523-5
7.
Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: Diagnosis, antimicrobial therapy, and management of complications: A scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435–86. doi:10.1161/CIR.0000000000000296
8.
Bai B, Hu K, Zeng J, et al. Linezolid consumption facilitates the development of linezolid resistance in Enterococcus faecalis in a tertiary-care hospital: A 5-year surveillance study. Microbial Drug Resistance. 2019;25(6):791–8. doi:10.1089/mdr.2018.0005
9.
Bains HS, Weinberg DV, Feder RS, et al. Postoperative vancomycin-resistant Enterococcus faecium endophthalmitis. Archives of Ophthalmology. 2007;125:1292–3. doi:10.1001/archopht.125.9.1292
10.
Balli EP, Venetis CA, Miyakis S. Systematic review and meta-analysis of linezolid versus daptomycin for treatment of vancomycin-resistant enterococcal bacteremia. Antimicrobial Agents and Chemotherapy. 2014;58:734–9. doi:10.1128/AAC.01289-13
11.
Bandı́n-Vilar E, Garcı́a-Quintanilla L, Castro-Balado A, et al. A review of population pharmacokinetic analyses of linezolid. Clinical Pharmacokinetics. 2022;61(6):789–817. doi:10.1007/s40262-022-01125-2
12.
Barnhill AE, Brewer MT, Carlson SA. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrobial Agents and Chemotherapy. 2012;56:4046–51. doi:10.1128/AAC.00678-12
13.
Bender JK, Cattoir V, Hegstad K, et al. Update on prevalence and mechanisms of resistance to linezolid, tigecycline, and daptomycin in enterococci in Europe: Towards a common nomenclature. Drug Resistance Updates. 2018;40:25–39. doi:10.1016/j.drup.2018.10.002
14.
Bernstein WB, Trotta RF, Rector JT, et al. Mechanisms for linezolid-induced anemia and thrombocytopenia. Annals of Pharmacotherapy. 2003;37:517–20. doi:10.1345/aph.1C361
15.
Biedenbach DJ, Jones RN. In vitro activity of linezolid (U-100766) against Haemophilus influenzae measured by three different susceptibility testing methods. Diagnostic Microbiology and Infectious Disease. 2001;39:49–53. doi:10.1016/s0732-8893(00)00216-1
16.
Birmingham MC, Rayner CR, Meagher AK, et al. Linezolid for the treatment of multidrug-resistant, gram-positive infections: Experience from a compassionate-use program. Clinical Infectious Diseases. 2003;36:159–68. doi:10.1086/345744
17.
Boak LM, Rayner CR, Grayson L, et al. Clinical population pharmacokinetics and toxicodynamics of linezolid. Antimicrobial Agents and Chemotherapy. 2014;58:2334–43. doi:10.1128/AAC.01885-13
18.
Bolhuis M, Altena R van, Uges DR, et al. Clarithromycin significantly increases linezolid serum concentrations. Antimicrobial Agents and Chemotherapy. 2010;54:5418–9. doi:10.1128/AAC.00757-10
19.
Bonilla H, Huband MD, Seidel J, et al. Multicity outbreak of linezolid-resistant Staphylococcus epidermidis associated with clonal spread of a cfr-containing strain. Clinical Infectious Diseases. 2010;51:796–800. doi:10.1086/656281
20.
Bradley JS, Antadze T, Ninov B, et al. Safety and efficacy of oral and/or intravenous tedizolid phosphate from a randomized phase 3 trial in adolescents with acute bacterial skin and skin structure infections. Pediatric Infectious Disease Journal. 2021;40:238–44. doi:10.1097/INF.0000000000002975
21.
Brenciani A, Morroni G, Schwarz S, et al. Oxazolidinones: Mechanisms of resistance and mobile genetic elements involved. Journal of Antimicrobial Chemotherapy. 2022;77(10):2596–621. doi:10.1093/jac/dkac263
22.
Britt NS, Potter EM, Patel N, et al. Comparison of the effectiveness and safety of linezolid and daptomycin in vancomycin-resistant enterococcal bloodstream infection: A national cohort study of Veterans Affairs patients. Clinical Infectious Diseases. 2015;61:871–8. doi:10.1093/cid/civ444
23.
Brown NM, Goodman AL, Horner C, et al. Treatment of methicillin-resistant Staphylococcus aureus (MRSA): Updated guidelines from the UK. JAC-Antimicrobial Resistance. 2021;3(1):dlaa114. doi:10.1093/jacamr/dlaa114
24.
Brown-Elliott BA, Crist DJ, Mann LB, et al. In vitro activity of linezolid against slowly growing nontuberculous mycobacteria. Antimicrobial Agents and Chemotherapy. 2003;47:1736–8. doi:10.1128/AAC.47.5.1736-1738.2003
25.
Brown-Elliott BA, Wallace Jr R. J. In vitro susceptibility testing of tedizolid against isolates of Nocardia. Antimicrobial Agents and Chemotherapy. 2017;61:e01537–17. doi:10.1128/AAC.01537-17
26.
Brown-Elliott BA, Wallace Jr R. J. In vitro susceptibility testing of tedizolid against non-tuberculous mycobacteria. Journal of Clinical Microbiology. 2017;55:1747–54. doi:10.1128/JCM.00274-17
27.
Burdette SD, Trotman R. Tedizolid: The first once-daily oxazolidinone class antibiotic. Clinical Infectious Diseases. 2015;61(8):1315–21. doi:10.1093/cid/civ501
28.
Carroll MW, Choi H, Min S, et al. Rhabdomyolysis in a patient treated with linezolid for extensively drug-resistant tuberculosis. Clinical Infectious Diseases. 2012;54(11):1624–7. doi:10.1093/cid/cis271
29.
Carvalhaes CG, Sader HS, Flamm RK, et al. Assessment of tedizolid in vitro activity and resistance mechanisms against a collection of Enterococcus spp. Causing invasive infections, including isolates requiring an optimized dosing strategy for daptomycin from U.S. And european medical centers, 2016 to 2018. Antimicrobial Agents and Chemotherapy. 2020;64:e00175–20. doi:10.1128/AAC.00175-20
30.
Carvalhaes CG, Sader HS, Streit JM, et al. Five-year analysis of the in vitro activity of tedizolid against a worldwide collection of indicated species causing clinical infections: Results from the surveillance of tedizolid activity and resistance (STAR) programme. JAC-Antimicrobial Resistance. 2022;4(5):dlac088. doi:10.1093/jacamr/dlac088
31.
Cattaneo D, Alffenaar JW, Neely M. Drug monitoring and individual dose optimization of antimicrobial drugs: oxazolidinones. Expert Opinion on Drug Metabolism & Toxicology. 2016;12:533–44. doi:10.1517/17425255.2016.1166204
32.
Cattaneo D, Gervasoni C, Cozzi V, et al. Therapeutic drug management of linezolid: A missed opportunity for clinicians? International Journal of Antimicrobial Agents. 2016;48(6):728–31. doi:10.1016/j.ijantimicag.2016.09.026
33.
Chen H, Du Y, Xia Q, et al. Role of linezolid combination therapy for serious infections: Review of the current evidence. European Journal of Clinical Microbiology & Infectious Diseases. 2020;39(6):1043–52. doi:10.1007/s10096-019-03801-x
34.
Chomei Y, Nishimura S, Iwata K. Long-term use of tedizolid for pulmonary nocardiosis. Journal of Infection and Chemotherapy. 2022;28(8):1172–6. doi:10.1016/j.jiac.2022.04.009
35.
Chuang YC, Wang JT, Lin HY, et al. Daptomycin versus linezolid for treatment of vancomycin-resistant enterococcal bacteremia: Systematic review and meta-analysis. BMC Infectious Diseases. 2014;14:687–97. doi:10.1186/s12879-014-0687-9
36.
Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. 32nd ed. Wayne, PA: CLSI; 2022.
37.
Colca JR, McDonald WG, Waldon DJ, et al. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. Journal of Biological Chemistry. 2003;278(24):21972–9. doi:10.1074/jbc.M302109200
38.
Conradie F, Bagdasaryan TR, Borisov S, et al. Bedaquiline-pretomanid-linezolid regimens for drug-resistant tuberculosis (ZeNix). New England Journal of Medicine. 2022;387(9):810–23. doi:10.1056/NEJMoa2119430
39.
Conradie F, Diacon AH, Ngubane N, et al. Treatment of highly drug-resistant pulmonary tuberculosis (Nix-TB). New England Journal of Medicine. 2020;382(10):893–902. doi:10.1056/NEJMoa1901814
40.
Corsini Campioli C, Castillo Almeida NE, O’Horo JC, et al. Clinical presentation, management, and outcomes of patients with brain abscess due to Nocardia spp. Open Forum Infectious Diseases. 2021;8(4):ofab067. doi:10.1093/ofid/ofab067
41.
Cortes-Penfield N, Ryder JH. Should linezolid replace clindamycin as the adjunctive antimicrobial of choice in group A streptococcal necrotizing soft tissue infection and toxic shock syndrome? A focused debate. Clinical Infectious Diseases. 2023;76(2):346–50. doi:10.1093/cid/ciac720
42.
Coussement J, Lebeaux D, Rouzaud C, et al. Nocardia infections in solid organ and hematopoietic stem cell transplant recipients. Current Opinion in Infectious Diseases. 2017;30(6):545–51. doi:10.1097/QCO.0000000000000404
43.
Cox HL, Sifri CD, Sawyer RG, et al. Institutional outbreak of linezolid-nonsusceptible Enterococcus faecium (LNEF) emerges during linezolid cycling in a surgical intensive care unit (SICU). 2012.
44.
Dagher M, Fowler Jr V. G., Wright PW, et al. A narrative review of early oral stepdown therapy for the treatment of uncomplicated Staphylococcus aureus bacteremia: Yay or nay? Open Forum Infectious Diseases. 2020;7(6):ofaa151. doi:10.1093/ofid/ofaa151
45.
Davidson N, Grigg MJ, Mcguinness SL, et al. Safety and outcomes of linezolid use for nocardiosis. Open Forum Infectious Diseases. 2020;7(4):ofaa090. doi:10.1093/ofid/ofaa090
46.
De Bus L, Depuydt P, Libbrecht L, et al. Severe drug-induced liver injury associated with prolonged use of linezolid. Journal of Medical Toxicology. 2010;6(3):322–6. doi:10.1007/s13181-010-0047-0
47.
De La Cruz O, Minces LR, Silveira FP. Experience with linezolid for the treatment of nocardiosis in organ transplant recipients. Journal of Infection. 2015;70(1):44–51. doi:10.1016/j.jinf.2014.08.010
48.
De Vriese AS, Van Coster R, Smet J, et al. Linezolid-induced inhibition of mitochondrial protein synthesis. Clinical Infectious Diseases. 2006;42:1111–7. doi:10.1086/501356
49.
Di Paolo A, Malacarne P, Guidotti E, et al. Pharmacological issues of linezolid: An updated critical review. Clinical Pharmacokinetics. 2010;49:439–47. doi:10.2165/11319960-000000000-00000
50.
Diaz L, Kiratisin P, Mendes RE, et al. Transferable plasmid-mediated resistance to linezolid due to cfr in a human clinical isolate of Enterococcus faecalis. Antimicrobial Agents and Chemotherapy. 2012;56:3917–22. doi:10.1128/AAC.00419-12
51.
Diekema D, Jones R. Oxazolidinone antibiotics. The Lancet. 2001;358(9297):1975–82. doi:10.1016/S0140-6736(01)06964-1
52.
Douros A, Grabowski K, Stahlmann R. Drug-drug interactions and safety of linezolid, tedizolid, and other oxazolidinones. Expert Opinion on Drug Metabolism & Toxicology. 2015;11:1849–59. doi:10.1517/17425255.2015.1098617
53.
Drusano GL, Liu W, Kulawy R, et al. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrobial Agents and Chemotherapy. 2011;55:5300–5. doi:10.1128/AAC.00502-11
54.
European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters, version 12.0 [Internet]. 2022. Available from: http://www.eucast.org
55.
Feng J, Xiang F, Cheng J, et al. Comparative efficacy and safety of vancomycin, linezolid, tedizolid, and daptomycin in treating patients with suspected or proved complicated skin and soft tissue infections: An updated network meta-analysis. Infectious Diseases and Therapy. 2021;10(3):1531–47. doi:10.1007/s40121-021-00456-0
56.
Ferry T, Conrad A, Senneville E, et al. Safety of tedizolid as suppressive antimicrobial therapy for patients with complex implant-associated bone and joint infection due to multidrug-resistant gram-positive pathogens: Results from the TediSAT cohort study. Open Forum Infectious Diseases. 2021;8(7):ofab351. doi:10.1093/ofid/ofab351
57.
Flanagan S, McKee EE, Das D, et al. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrobial Agents and Chemotherapy. 2015;59:178–85. doi:10.1128/AAC.03684-14
58.
Foti C, Piperno A, Scala A, et al. Oxazolidinone antibiotics: Chemical, biological, and analytical aspects. Molecules. 2021;26(14):4280. doi:10.3390/molecules26144280
59.
Frasca KL, Schuster MG. Vancomycin-resistant enterococcal meningitis in an autologous stem cell transplant recipient cured with linezolid. Transplant Infectious Disease. 2013;15:E1–4. doi:10.1111/tid.12035
60.
Gandelman K, Zhu T, Fahmi OA, et al. Unexpected effect of rifampin on the pharmacokinetics of linezolid: In silico and in vitro approaches to explain its mechanism. Journal of Clinical Pharmacology. 2011;51:229–36. doi:10.1177/0091270010366445
61.
Garcı́a MS, De la Torre MA, Morales G, et al. Clinical outbreak of linezolid-resistant Staphylococcus aureus in an intensive care unit. JAMA. 2010;303:2260–4. doi:10.1001/jama.2010.757
62.
Gatti M, Fusaroli M, Raschi E, et al. Serious adverse events with tedizolid and linezolid: Pharmacovigilance insights through the FDA adverse event reporting system. Expert Opinion on Drug Safety. 2021;20(11):1421–31. doi:10.1080/14740338.2021.1956461
63.
Gatti M, Raschi E, De Ponti F. Serotonin syndrome by drug interactions with linezolid: Clues from pharmacovigilance-pharmacokinetic/pharmacodynamics analysis. European Journal of Clinical Pharmacology. 2021;77(2):233–9. doi:10.1007/s00228-020-02990-1
64.
Gerson SL, Kaplan SL, Bruss JB, et al. Hematologic effects of linezolid: Summary of clinical experience. Antimicrobial Agents and Chemotherapy. 2002;46:2723–6. doi:10.1128/AAC.46.8.2723-2726.2002
65.
Goldstein EJ, Citron DM, Merriam CV. Linezolid activity compared to those of selected macrolides and other agents against aerobic and anaerobic pathogens isolated from soft tissue bite infections in humans. Antimicrobial Agents and Chemotherapy. 1999;43:1469–74. doi:10.1128/AAC.43.6.1469
66.
Grim SA, Rene L, Gupta S, et al. Safety of linezolid in patients with baseline thrombocytopenia. Journal of Antimicrobial Chemotherapy. 2008;62:850–1. doi:10.1093/jac/dkn254
67.
Hamdi AM, Fida M, Deml SM, et al. Retrospective analysis of antimicrobial susceptibility profiles of Nocardia species from a tertiary hospital and reference laboratory, 2011 to 2017. Antimicrobial Agents and Chemotherapy. 2020;64(3):e01868–19. doi:10.1128/AAC.01868-19
68.
Hölzel CS, Harms KS, Schwaiger K, et al. Resistance to linezolid in a porcine Clostridium perfringens strain carrying a mutation in the rplD gene encoding the ribosomal protein L4. Antimicrobial Agents and Chemotherapy. 2010;54:1351–3. doi:10.1128/AAC.01208-09
69.
Huerga H, Khan U, Bastard M, et al. Safety and effectiveness outcomes from a 14-country cohort of patients with multi-drug resistant tuberculosis treated concomitantly with bedaquiline, delamanid, and other second-line drugs. Clinical Infectious Diseases. 2022;75(8):1307–14. doi:10.1093/cid/ciac176
70.
Imperial MZ, Nedelman JR, Conradie F, et al. Proposed linezolid dosing strategies to minimize adverse events for treatment of extensively drug-resistant tuberculosis. Clinical Infectious Diseases. 2022;74(10):1736–47. doi:10.1093/cid/ciab699
71.
Iqbal K, Milioudi A, Wicha SG. Pharmacokinetics and pharmacodynamics of tedizolid. Clinical Pharmacokinetics. 2022;61(4):489–503. doi:10.1007/s40262-021-01099-7
72.
Iqbal K, Rohde H, Huang J, et al. A pharmacokinetic-pharmacodynamic model-based analysis of tedizolid against enterococci using the hollow-fibre infection model. Journal of Antimicrobial Chemotherapy. 2022;77(9):2470–8. doi:10.1093/jac/dkac208
73.
Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. New England Journal of Medicine. 2019;380(5):415–24. doi:10.1056/NEJMoa1808312
74.
Jaksic B, Martinelli G, Perez-Oteyza J, et al. Efficacy and safety of linezolid compared with vancomycin in a randomized, double-blind study of febrile neutropenic patients with cancer. Clinical Infectious Diseases. 2006;42:597–607. doi:10.1086/500139
75.
Jang HC, Kim S-H, Kim KH, et al. Salvage treatment for persistent methicillin-resistant Staphylococcus aureus bacteremia: Efficacy of linezolid with or without carbapenem. Clinical Infectious Diseases. 2009;49:395–401. doi:10.1086/600295
76.
Jaspard M, Butel N, El Helali N, et al. Linezolid-associated neurologic adverse effects in patients with multidrug-resistant tuberculosis, France. Emerging Infectious Diseases. 2020;26(8):1792–800. doi:10.3201/eid2608.191499
77.
Kalil AC, Klompas M, Haynatzki G, et al. Treatment of hospital-acquired pneumonia with linezolid or vancomycin: A systematic review and meta-analysis. BMJ Open. 2013;3(10):e003912. doi:10.1136/bmjopen-2013-003912
78.
Kaplan SL, Patterson L, Edwards KM, et al. Linezolid for the treatment of community-acquired pneumonia in hospitalized children. Pediatric Infectious Disease Journal. 2001;20(5):488–94. doi:10.1097/00006454-200105000-00004
79.
Kim DH, Kim S-Y, Koh W-J, et al. In vitro activity of oxazolidinone against nontuberculous mycobacteria, including macrolide-resistant clinical isolates. Antimicrobial Agents and Chemotherapy. 2021;65(7):e02306–20. doi:10.1128/AAC.02306-20
80.
Koch BCP, Zhao Q, Oosterhoff M, et al. The mysteries of target site concentrations of antibiotics in bone and joint infections: What is known? A narrative review. Expert Opinion on Drug Metabolism & Toxicology. 2022;18(9):587–600. doi:10.1080/17425255.2022.2122813
81.
Kullar R, Sakoulas G, Deresinski S, et al. When sepsis persists: A review of MRSA bacteraemia salvage therapy. Journal of Antimicrobial Chemotherapy. 2016;71:576–86. doi:10.1093/jac/dkv368
82.
Larruskain J, Idigoras P, Marimón JM, et al. Susceptibility of 186 Nocardia sp. Isolates to 20 antimicrobial agents. Antimicrobial Agents and Chemotherapy. 2011;55(6):2995–8. doi:10.1128/AAC.01279-10
83.
Lemaire S, Tulkens PM, Van Bambeke F. Cellular pharmacokinetics of the novel biaryloxazolidinone radezolid in phagocytic cells: Studies with macrophages and polymorphonuclear neutrophils. Antimicrobial Agents and Chemotherapy. 2010;54(6):2540–8. doi:10.1128/AAC.01723-09
84.
Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clinical Infectious Diseases. 2011;52:1–38. doi:10.1093/cid/ciq146
85.
Locke JB, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrobial Agents and Chemotherapy. 2010;54(12):5337–43. doi:10.1128/AAC.00663-10
86.
Locke JB, Zurenko GE, Shaw KJ, et al. Tedizolid for the management of human infections: In vitro characteristics. Clinical Infectious Diseases. 2014;58(Suppl 1):S35–42. doi:10.1093/cid/cit616
87.
Long KS, Vester B. Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrobial Agents and Chemotherapy. 2012;56:603–12. doi:10.1128/AAC.05702-11
88.
Luque S, Grau S, Alvarez-Lerma F, et al. Plasma and cerebrospinal fluid concentrations of linezolid in neurosurgical critically ill patients with proven or suspected central nervous system infections. International Journal of Antimicrobial Agents. 2014;44(5):409–15. doi:10.1016/j.ijantimicag.2014.07.001
89.
Luque S, Munoz-Bermudez R, Echeverria-Esnal D, et al. Linezolid dosing in patients with liver cirrhosis: Standard dosing risk toxicity. Therapeutic Drug Monitoring. 2019;41(6):732–9. doi:10.1097/FTD.0000000000000665
90.
Lv X, Alder J, Li L, et al. Efficacy and safety of tedizolid phosphate versus linezolid in a randomized phase 3 trial in patients with acute bacterial skin and skin structure infection. Antimicrobial Agents and Chemotherapy. 2019;63(7):e02252–18. doi:10.1128/AAC.02252-18
91.
Maltempe FG, Caleffi-Ferracioli KR, Amaral RCR do, et al. Activity of rifampin and linezolid combination in Mycobacterium tuberculosis. Tuberculosis. 2017;104:24–9. doi:10.1016/j.tube.2017.02.006
92.
Mancino P, Ucciferri C, Falasca K, et al. Methicillin-resistant Staphylococcus epidermidis (MRSE) endocarditis treated with linezolid. Scandinavian Journal of Infectious Diseases. 2008;40:67–73. doi:10.1080/00365540701522496
93.
Margalit I, Lebeaux D, Tishler O, et al. How do i manage nocardiosis? Clinical Microbiology and Infection. 2021;27(4):550–8. doi:10.1016/j.cmi.2020.12.019
94.
Matin A, Sharma S, Mathur P, et al. Myelosuppression-sparing treatment of central nervous system nocardiosis in a multiple myeloma patient utilizing a tedizolid-based regimen: A case report. International Journal of Antimicrobial Agents. 2017;49:488–92. doi:10.1016/j.ijantimicag.2016.11.014
95.
Matsumoto K, Takeshita A, Ikawa K, et al. Higher linezolid exposure and higher frequency of thrombocytopenia in patients with renal dysfunction. International Journal of Antimicrobial Agents. 2010;36:179–81. doi:10.1016/j.ijantimicag.2010.02.019
96.
McCool R, Gould IM, Eales J, et al. Systematic review and network meta-analysis of tedizolid for the treatment of acute bacterial skin and skin-structure infections caused by MRSA. BMC Infectious Diseases. 2017;17:39–51. doi:10.1186/s12879-016-2128-4
97.
McKinnell JA, Arias CA. Linezolid vs daptomycin for vancomycin-resistant enterococci: The evidence gap between trials and clinical experience. Clinical Infectious Diseases. 2015;61:879–82. doi:10.1093/cid/civ449
98.
McNally MA, Farooq S, Tschen J, et al. Linezolid as a treatment option for cutaneous non-tuberculous mycobacterial infections. Dermatology Online Journal. 2020;26(9):8.
99.
McNeil SA, Clark NM, Chandrasekar PH, et al. Successful treatment of vancomycin-resistant Enterococcus faecium bacteremia with linezolid after failure of treatment with Synercid (quinupristin/dalfopristin). Clinical Infectious Diseases. 2000;30:403–4. doi:10.1086/313654
Mendes RE, Deshpande L, Streit JM, et al.ZAAPS programme results for 2016: An activity and spectrum analysis of linezolid using clinical isolates from medical centres in 42 countries. Journal of Antimicrobial Chemotherapy. 2018;73(7):1880–7. doi:10.1093/jac/dky099
102.
Mensa Vendrell M, Tasias Pitarch M, Salavert Lleti M, et al. Safety and tolerability of more than six days of tedizolid treatment. Antimicrobial Agents and Chemotherapy. 2020;64(7):e00356–20. doi:10.1128/AAC.00356-20
Mikamo H, Takesue Y, Iwamoto Y, et al. Efficacy, safety and pharmacokinetics of tedizolid versus linezolid in patients with skin and soft tissue infections in Japan — results of a randomized, multicenter phase 3 study. Journal of Infection and Chemotherapy. 2018;24(6):434–42. doi:10.1016/j.jiac.2018.01.010
105.
Mirzayev F, Viney K, Linh NN, et al.World Health Organization recommendations on the treatment of drug-resistant tuberculosis, 2020 update. European Respiratory Journal. 2021;57(6):2003300. doi:10.1183/13993003.03300-2020
106.
Moellering RC. Linezolid: The first oxazolidinone antimicrobial. Annals of Internal Medicine. 2003;138(2):135–42. doi:10.7326/0003-4819-138-2-200301210-00015
107.
Moran GJ, Fang E, Corey GR, et al. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): A randomised, double-blind, phase 3, non-inferiority trial. The Lancet Infectious Diseases. 2014;14:696–705. doi:10.1016/S1473-3099(14)70737-6
108.
Morrisette T, Molina KC, Da Silva B, et al. Real-world use of tedizolid phosphate for 28 days or more: A case series describing tolerability and clinical success. Open Forum Infectious Diseases. 2022;9(6):ofac028. doi:10.1093/ofid/ofac028
109.
Munoz P, De la Villa S, Martı́nez-Sellés M, et al. Linezolid for infective endocarditis: A structured approach based on a national database experience. Medicine (Baltimore). 2021;100(51):e27597. doi:10.1097/MD.0000000000027597
110.
Nagel S, Köhrmann M, Huttner HB, et al. Linezolid-induced posterior reversible leukoencephalopathy syndrome. Archives of Neurology. 2007;64:746–8. doi:10.1001/archneur.64.5.746
111.
Narita M, Tsuji BT, Yu VL. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy. 2007;27:1189–97. doi:10.1592/phco.27.8.1189
112.
Ntokou E, Stathopoulos C, Kristo I, et al. Intensive care unit dissemination of multiple clones of linezolid-resistant Enterococcus faecalis and Enterococcus faecium. Journal of Antimicrobial Chemotherapy. 2012;67:1819–23. doi:10.1093/jac/dks146
113.
Ntziora F, Falagas ME. Linezolid for the treatment of patients with central nervous system infection. Annals of Pharmacotherapy. 2007;41:296–308. doi:10.1345/aph.1H307
114.
Obach RS. Linezolid metabolism is catalyzed by cytochrome P450 2J2, 4F2, and 1B1. Drug Metabolism and Disposition. 2022;50(4):413–21. doi:10.1124/dmd.121.000776
115.
Pea F, Viale P, Cojutti P, et al. Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. Journal of Antimicrobial Chemotherapy. 2012;67:2034–42. doi:10.1093/jac/dks153
116.
Pharmacia and Upjohn Company. Zyvox® (linezolid) [Package Insert]. New York, NY; 2021.
117.
Pogue JM, Paterson DL, Pasculle W, et al. Determination of risk factors associated with isolation of linezolid-resistant strains of vancomycin-resistant Enterococcus. Infection Control and Hospital Epidemiology. 2007;28:1382–8. doi:10.1086/523276
118.
Poon YK, La Hoz RM, Hynan LS, et al. Tedizolid vs linezolid for the treatment of nontuberculous mycobacteria infections in solid organ transplant recipients. Open Forum Infectious Diseases. 2021;8(4):ofab093. doi:10.1093/ofid/ofab093
119.
Powers JH, Lin D, Ross D, et al.FDA evaluation of antimicrobials: Subgroup analysis. Chest. 2004;127:2298–9. doi:10.1378/chest.127.6.2298
120.
Prokocimer P, De Anda C, Fang E, et al. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: The ESTABLISH-1 randomized trial. JAMA. 2013;309:559–69. doi:10.1001/jama.2013.241
121.
Rao GG, Konicki R, Cattaneo D, et al. Therapeutic drug monitoring can improve linezolid dosing regimens in current clinical practice: A review of linezolid pharmacokinetics and pharmacodynamics. Therapeutic Drug Monitoring. 2020;42(1):83–92. doi:10.1097/FTD.0000000000000710
122.
Rao N, Hamilton CW. Efficacy and safety of linezolid for gram-positive orthopedic infections: A prospective case series. Diagnostic Microbiology and Infectious Disease. 2007;59:173–9. doi:10.1016/j.diagmicrobio.2007.04.006
123.
Refaat M, Hyle E, Malhotra R, et al. Linezolid-induced lingua villosa nigra. American Journal of Medicine. 2008;121:e1. doi:10.1016/j.amjmed.2008.06.039
124.
Roger C, Roberts JA, Muller L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clinical Pharmacokinetics. 2018;57:559–75. doi:10.1007/s40262-017-0601-x
125.
Ruiz P, Causse M, Vaquero M, et al. In vitro activity of tedizolid against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2019;63(4):e01939–18. doi:10.1128/AAC.01939-18
126.
Rybak JM, Roberts K. Tedizolid phosphate: A next-generation oxazolidinone. Infectious Diseases and Therapy. 2015;4(1):1–14. doi:10.1007/s40121-015-0060-3
127.
Sakai Y, Naito T, Arima C, et al. Potential drug interaction between warfarin and linezolid. Internal Medicine. 2015;54:459–64. doi:10.2169/internalmedicine.54.3146
128.
San Pedro GS, Cammarata SK, Oliphant TH, et al. Linezolid versus ceftriaxone/cefpodoxime in patients hospitalized for the treatment of Streptococcus pneumoniae pneumonia. Scandinavian Journal of Infectious Diseases. 2002;34(10):720–8. doi:10.1080/00365540210147525
129.
Santini A, Ronchi D, Garbellini M, et al. Linezolid-induced lactic acidosis: The thin line between bacterial and mitochondrial ribosomes. Expert Opinion on Drug Safety. 2017;16:833–43. doi:10.1080/14740338.2017.1335305
130.
Scheetz MH, Knechtel SA, Malczynski M, et al. Increasing incidence of linezolid-intermediate or -resistant vancomycin-resistant Enterococcus faecium strains parallels increasing linezolid consumption. Antimicrobial Agents and Chemotherapy. 2008;52:2256–9. doi:10.1128/AAC.00070-08
131.
Shaikh ZH, Peloquin CA, Ericsson CD. Successful treatment of vancomycin-resistant Enterococcus faecium meningitis with linezolid: Case report and literature review. Scandinavian Journal of Infectious Diseases. 2001;33:375–9. doi:10.1080/003655401750174048
132.
Shaw TD, Smyth M, Turner G, et al. Prolonged tedizolid use in cutaneous non-tuberculous mycobacterial infection. Journal of Clinical Tuberculosis and Other Mycobacterial Diseases. 2021;24:100261. doi:10.1016/j.jctube.2021.100261
133.
Shi C, Jin W, Xie Y, et al. Efficacy and safety of daptomycin versus linezolid treatment in patients with vancomycin-resistant enterococcal bacteraemia: An updated systematic review and meta-analysis. Journal of Global Antimicrobial Resistance. 2020;21:235–45. doi:10.1016/j.jgar.2019.10.008
134.
Shoen C, DeStefano M, Hafkin B, et al. In vitro and in vivo activities of contezolid (MRX-I) against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2018;62(8):e00493–18. doi:10.1128/AAC.00493-18
135.
Shorr AF, Lodise TP, Corey GR, et al. Analysis of the phase 3 ESTABLISH trials of tedizolid versus linezolid in acute bacterial skin and skin structure infections. Antimicrobial Agents and Chemotherapy. 2015;59:864–71. doi:10.1128/AAC.03688-14
136.
Si S, Durkin MJ, Mercier MM, et al. Successful treatment of prosthetic joint infection due to vancomycin-resistant enterococci with tedizolid. Infectious Diseases in Clinical Practice (Baltimore Md). 2017;25:105–7. doi:10.1097/IPC.0000000000000466
137.
Slee AM, Wuonola MA, McRipley RJ, et al. Oxazolidinones, a new class of synthetic antibacterial agents: In vitro and in vivo activities of DuP 105 and DuP 721. Antimicrobial Agents and Chemotherapy. 1987;31(11):1791–7. doi:10.1128/AAC.31.11.1791
138.
Soriano A, Gómez J, Gómez L, et al. Efficacy and tolerability of prolonged linezolid therapy in the treatment of orthopedic implant infections. European Journal of Clinical Microbiology & Infectious Diseases. 2007;26:353–6. doi:10.1007/s10096-007-0289-1
139.
Soueges S, Triffault-Fillit C, Roux S, et al. Long-term use of liposomal nebulized amikacin and tedizolid for the treatment of disseminated nocardiosis after allogeneic hematopoietic stem cell transplantation. European Journal of Clinical Microbiology & Infectious Diseases. 2021;40(9):2033–6. doi:10.1007/s10096-021-04204-7
140.
Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clinical Infectious Diseases. 2014;59(2):e10–52. doi:10.1093/cid/ciu444
141.
Sudhindra P, Lee L, Wang G, et al. Tedizolid for treatment of enterococcal bacteremia. Open Forum Infectious Diseases. 2016;3(suppl 1):1–2. doi:10.1093/ofid/ofw194.16
142.
Tornheim JA, Intini E, Gupta A, et al. Clinical features associated with linezolid resistance among multidrug resistant tuberculosis patients at a tertiary care hospital in mumbai, india. Journal of Clinical Tuberculosis and Other Mycobacterial Diseases. 2020;20:100175. doi:10.1016/j.jctube.2020.100175
143.
Traver EC, Heil EL, Schmalzle SA. A cross-sectional analysis of linezolid in combination with methadone or buprenorphine as a cause of serotonin toxicity. Open Forum Infectious Diseases. 2022;9(7):ofac331. doi:10.1093/ofid/ofac331
144.
Tsigrelis C, Singh KV, Coutinho TD, et al. Vancomycin-resistant Enterococcus faecalis endocarditis: Linezolid failure and strain characterization of virulence factors. Journal of Clinical Microbiology. 2007;45:631–5. doi:10.1128/JCM.02152-06
145.
Velez JC, Janech MG. A case of lactic acidosis induced by linezolid. Nature Reviews Nephrology. 2010;6:236–42. doi:10.1038/nrneph.2010.20
146.
Villani P, Regazzi MG, Marubbi F, et al. Cerebrospinal fluid linezolid concentrations in postneurosurgical central nervous system infections. Antimicrobial Agents and Chemotherapy. 2002;46:936–7. doi:10.1128/AAC.46.3.936-937.2002
147.
Viswanathan P, Iarikov D, Wassel R, et al. Hypoglycemia in patients treated with linezolid. Clinical Infectious Diseases. 2014;59:e93–5. doi:10.1093/cid/ciu487
148.
Wald-Dickler N, Holtom P, Spellberg B. Busting the myth of “static vs cidal”: A systematic literature review. Clinical Infectious Diseases. 2018;66(9):1470–4. doi:10.1093/cid/cix1127
149.
Wallace RJ, Brown-Elliott BA, Ward SC, et al. Activities of linezolid against rapidly growing mycobacteria. Antimicrobial Agents and Chemotherapy. 2001;45:764–7. doi:10.1128/AAC.45.3.764-767.2001
150.
Wang S, Cai C, Shen Y, et al. In vitro activity of contezolid against methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus, and strains with linezolid resistance genes from china. Frontiers in Microbiology. 2021;12:729900. doi:10.3389/fmicb.2021.729900
151.
Wang Y, Lv Y, Cai J, et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. Journal of Antimicrobial Chemotherapy. 2015;70:2182–90. doi:10.1093/jac/dkv116
152.
Wang Y, Zou Y, Xie J, et al. Linezolid versus vancomycin for the treatment of suspected methicillin-resistant Staphylococcus aureus nosocomial pneumonia: A systematic review employing meta-analysis. European Journal of Clinical Pharmacology. 2015;71:107–15. doi:10.1007/s00228-014-1775-x
153.
Watkins RR, Lemonovich TL, File TH. An evidence-based review of linezolid for the treatment of methicillin-resistant Staphylococcus aureus (MRSA): Place in therapy. Core Evidence. 2012;7:131–43. doi:10.2147/CE.S33474
154.
Webster DP, Griffiths S, Bowler IC. Failure of linezolid therapy for post-neurosurgical meningitis due to Enterococcus faecium. Journal of Antimicrobial Chemotherapy. 2009;63:622–3. doi:10.1093/jac/dkn525
155.
Wen S, Gao X, Zhao W, et al. Comparison of the in vitro activity of linezolid, tedizolid, sutezolid, and delpazolid against rapidly growing mycobacteria isolated in beijing, china. International Journal of Infectious Diseases. 2021;109:253–60. doi:10.1016/j.ijid.2021.06.055
156.
Wilcox MH, Tack KJ, Bouza E, et al. Complicated skin and skin-structure infections and catheter-related bloodstream infections: Noninferiority of linezolid in a phase 3 study. Clinical Infectious Diseases. 2009;48:203–12. doi:10.1086/595686
157.
Willekens R, Puig-Asensio M, Ruiz-Camps I, et al. Early oral switch to linezolid for low-risk patients with Staphylococcus aureus bloodstream infections: A propensity-matched cohort study. Clinical Infectious Diseases. 2019;69(3):381–7. doi:10.1093/cid/ciy916
158.
Wunderink RG, Niederman MS, Kollef MH, et al. Linezolid in methicillin-resistant Staphylococcus aureus nosocomial pneumonia: A randomized, controlled study. Clinical Infectious Diseases. 2012;54:621–9. doi:10.1093/cid/cir895
159.
Wunderink RG, Rello J, Cammarata SK, et al. Linezolid vs vancomycin: Analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest. 2003;124:1789–97. doi:10.1378/chest.124.5.1789
160.
Wunderink RG, Roquilly A, Croce M, et al. A phase 3 randomized, double-blind study comparing tedizolid phosphate and linezolid for treatment of ventilated gram-positive hospital-acquired or ventilator-associated bacterial pneumonia. Clinical Infectious Diseases. 2021;73(3):e710–8. doi:10.1093/cid/ciab032
161.
Xiao J, Gill C, Liang L, et al. Use of translational pharmacokinetic/pharmacodynamic infection models to understand the impact of neutropenia on the efficacy of tedizolid phosphate. Antimicrobial Agents and Chemotherapy. 2019;63(1):e00822–18. doi:10.1128/AAC.00822-18
162.
Yang C, Lei H, Wang D, et al. In vitro activity of linezolid against clinical isolates of Mycobacterium tuberculosis, including multidrug-resistant and extensively drug-resistant strains from beijing, china. Japanese Journal of Infectious Diseases. 2012;65:240–2. doi:10.7883/yoken.65.240
163.
Yang JW, Kim YS, Choi SO, et al. Successful use of intravenous linezolid in CAPD patient with vancomycin-resistant enterococcal peritonitis. Peritoneal Dialysis International. 2011;31:209–10. doi:10.3747/pdi.2010.00130
164.
Yeager SD, Oliver JE, Shorman MA, et al. Comparison of linezolid step-down therapy to standard parenteral therapy in methicillin-resistant Staphylococcus aureus bloodstream infections. International Journal of Antimicrobial Agents. 2021;57(5):106329. doi:10.1016/j.ijantimicag.2021.106329
165.
Yeoh K, Globan M, Naimo P, et al. Identification and antimicrobial susceptibility of referred Nocardia isolates in victoria, australia 2009–2019. Journal of Medical Microbiology. 2022;71(8). doi:10.1099/jmm.0.001581
166.
Yu X, Huo F, Wang F, et al. In vitro antimicrobial activity comparison of linezolid, tedizolid, sutezolid and delpazolid against slowly growing mycobacteria isolated in beijing, china. Infection and Drug Resistance. 2021;14:4689–97. doi:10.2147/IDR.S336948
167.
Yue J, Dong BR, Yang M, et al. Linezolid versus vancomycin for skin and soft tissue infections. Cochrane Database of Systematic Reviews. 2016;1–42. doi:10.1002/14651858.CD008056.pub3
168.
Zeana C, Kubin CJ, Della-Latta P, et al. Vancomycin-resistant Enterococcus faecium meningitis successfully managed with linezolid: Case report and review of the literature. Clinical Infectious Diseases. 2001;33:477–82. doi:10.1086/321896
169.
Zhanel GG, Love R, Adam H, et al. Tedizolid: A novel oxazolidinone with potent activity against multidrug-resistant gram-positive pathogens. Drugs. 2015;75(3):253–70. doi:10.1007/s40265-015-0352-7
170.
Zhao W, Zheng M, Wang B, et al. Interactions of linezolid and second-line anti-tuberculous agents against multidrug-resistant Mycobacterium tuberculosis in vitro and in vivo. International Journal of Infectious Diseases. 2016;52:23–8. doi:10.1016/j.ijid.2016.09.011