Glycopeptides (Vancomycin and Teicoplanin) and Lipoglycopeptides (Telavancin, Oritavancin, and Dalbavancin)
Lecture Companion — Postgraduate Infectious Diseases
1 Introduction
The glycopeptides — vancomycin and teicoplanin — and the semi-synthetic lipoglycopeptides — telavancin, dalbavancin, and oritavancin — share a common mechanism of action: inhibition of late-stage peptidoglycan synthesis through high-affinity binding to the D-alanyl-D-alanine terminus of cell-wall precursors in dividing gram-positive organisms. Despite this shared mechanism, the five drugs differ markedly in their pharmacokinetic profiles, spectrum nuances, resistance liabilities, and approved clinical indications.
Vancomycin remains the backbone agent for serious methicillin-resistant Staphylococcus aureus (MRSA) infections worldwide, but its narrow therapeutic index, slow bactericidal activity, and the appearance of strains with reduced susceptibility — vancomycin-intermediate S. aureus (VISA), heteroresistant VISA (hVISA), and rare vancomycin-resistant S. aureus (VRSA) — have driven both pharmacokinetic refinement (AUC-guided dosing) and the development of the lipoglycopeptide successors (23,31,49).
- Vancomycin: backbone MRSA therapy; AUC-guided dosing now preferred over trough monitoring for serious MRSA infections (47).
- Teicoplanin: available in Europe/Asia; long half-life permits once-daily dosing after loading; possibly less nephrotoxic than vancomycin (53).
- Telavancin: dual-mechanism lipoglycopeptide; reserved for cSSSI and hospital-acquired/ventilator-associated pneumonia when alternatives are inappropriate; nephrotoxicity and QTc concerns.
- Dalbavancin: very long half-life (~14 days); single- or two-dose regimens for ABSSSI; emerging role in osteomyelitis and infective endocarditis.
- Oritavancin: terminal half-life ~245 hours; one-time single-dose ABSSSI therapy; interferes with coagulation assays.
2 Vancomycin
2.1 History
Vancomycin, the first glycopeptide antibiotic developed for clinical use, was isolated from Amycolatopsis orientalis (previously Streptomyces orientalis, later Nocardia orientalis) in a soil sample from Borneo in the mid-1950s. In 1958 vancomycin was introduced into clinical practice as an agent active against penicillin-resistant S. aureus. Use was quickly relegated to β-lactam–allergic patients after methicillin and cephalothin became available and because early lots — known as “Mississippi mud” for the colour imparted by impurities — were associated with substantial toxicity. Manufacturing purity improved markedly thereafter (31).
Since the 1980s vancomycin use has risen steadily — from approximately 2,000 kg/year in 1984 to over 11,200 kg/year in 1996 in the United States alone, paralleled in many other regions (15,31,32).

2.2 Chemistry
Vancomycin is a tricyclic glycopeptide of molecular weight ~1,449 Da consisting of a heptapeptide core with two attached sugars (vancosamine and glucose). The peptide backbone binds the terminal D-alanyl-D-alanine of nascent peptidoglycan via five hydrogen bonds, providing the high-affinity binding that defines class activity — but also its weakness, since a single mutation in the target can eliminate activity.
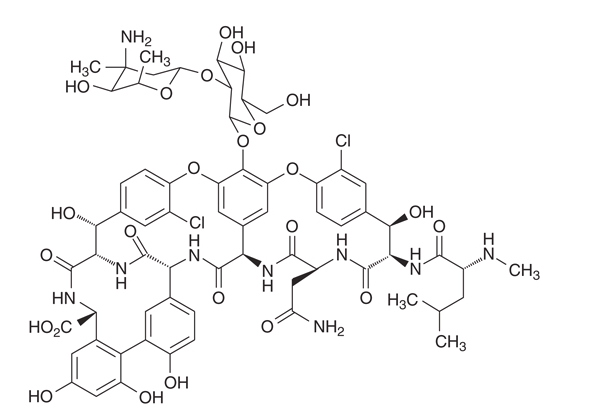
2.3 Mechanism of Action
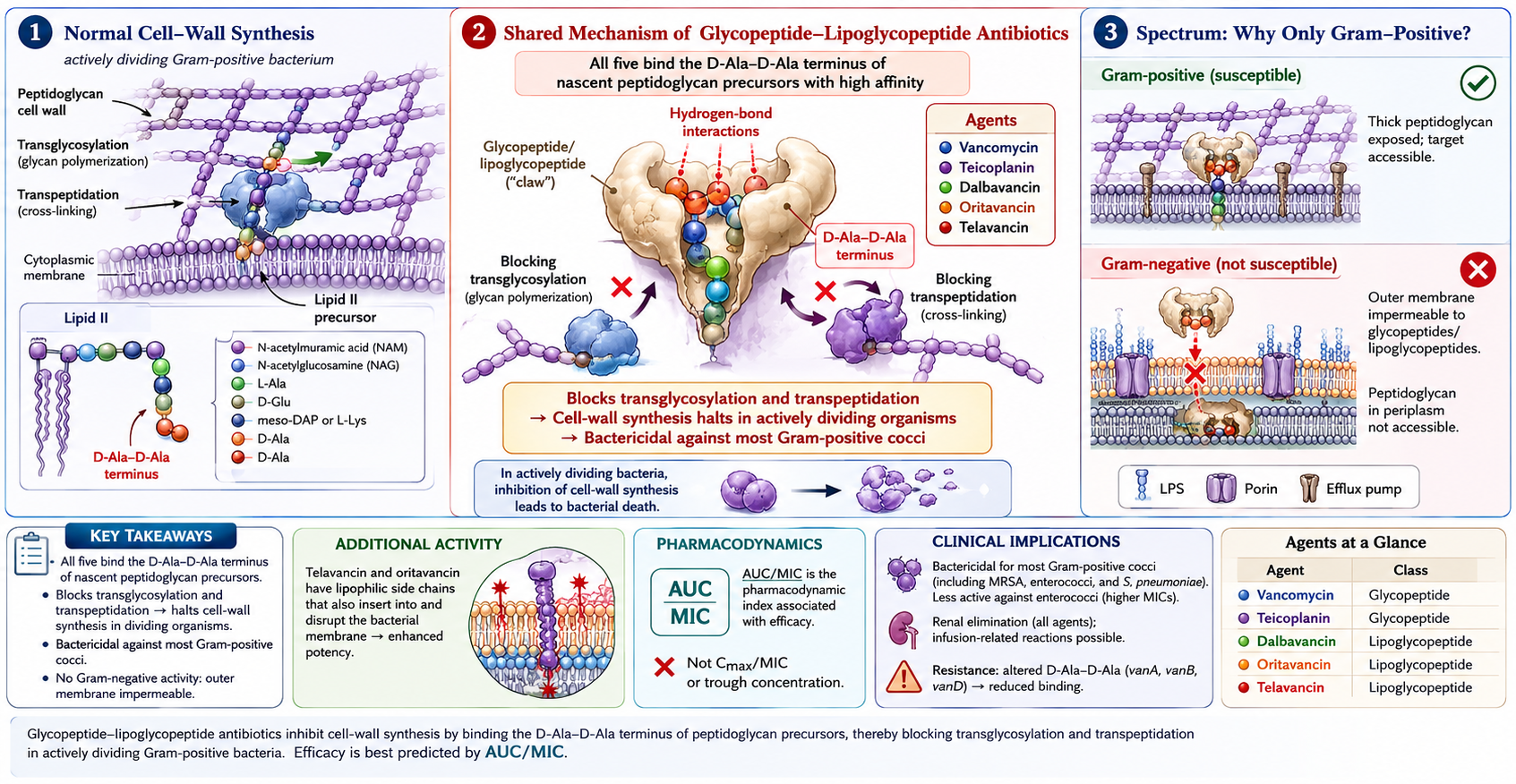
Vancomycin inhibits the late stages of cell-wall biosynthesis in dividing gram-positive organisms by complexing with the D-Ala-D-Ala C-terminus of peptidoglycan precursors, blocking both transglycosylation and transpeptidation (Figure 3). The drug does not penetrate the outer membrane of gram-negative organisms, accounting for its narrow spectrum. Glycopeptides are bactericidal but slowly so compared to β-lactams; the AUC/MIC index reflects this — duration of exposure matters more than peak concentration (44).
2.4 Antimicrobial Activity and Spectrum
Vancomycin is bactericidal against most gram-positive organisms, including Staphylococcus aureus (both methicillin-susceptible and -resistant), coagulase-negative staphylococci, streptococci (groups A–G, Streptococcus pneumoniae, viridans group), enterococci (when susceptible), Listeria monocytogenes, Corynebacterium jeikeium, Bacillus species, and gram-positive anaerobes including Clostridioides difficile and Cutibacterium acnes. Activity is concentration-independent and time-dependent, with the AUC/MIC ratio (rather than peak or trough alone) being the pharmacodynamic index that best predicts efficacy (47).
2.5 Resistance
2.5.1 Vancomycin-Resistant Enterococci (VRE)
Glycopeptide resistance in enterococci is mediated by van gene clusters that replace the terminal D-Ala-D-Ala of peptidoglycan precursors with D-Ala-D-lactate (VanA, VanB, VanD, VanM phenotypes — high-level resistance to vancomycin; VanB additionally retains teicoplanin susceptibility) or with D-Ala-D-serine (VanC, VanE, VanG, VanL, VanN — low-level resistance) (1,18). The 1000-fold affinity loss from D-Ala-D-lactate substitution — the removal of a single hydrogen bond from five — is one of the most elegant molecular explanations in microbiology (58,62). VanA and VanB phenotypes dominate clinically, most commonly in Enterococcus faecium (10,16). Surveillance data from the United States (NHSN), Europe (EARS-Net), and Latin America confirm continued global circulation and increasing prevalence in many regions (Figure 4) (14,61).
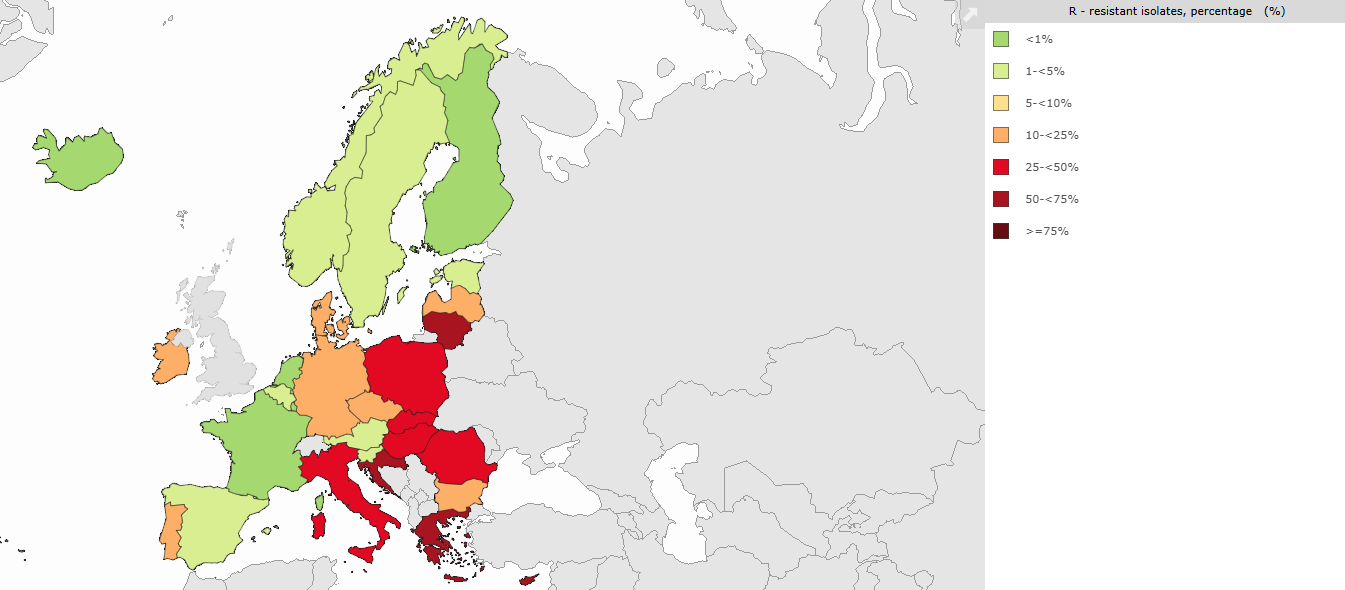
VanB is the phenotype to remember clinically: vancomycin-resistant but teicoplanin-susceptible, because the VanB regulatory system does not induce expression in response to teicoplanin. In regions with predominantly VanB phenotype, teicoplanin remains useful for enterococcal infections.
2.5.2 Vancomycin-Dependent Enterococci (VDE)
Rarely, enterococci that require vancomycin to grow have been described (Figure 5). The mechanism involves loss of the endogenous D-Ala-D-Ala ligase, leaving the organism entirely reliant on the vancomycin-induced D-Ala-D-Lac ligase. VDE strains are undetectable on routine media without vancomycin and can revert to vancomycin-independent growth. Though mostly a microbiological curiosity, VDE should be considered when growth occurs only on vancomycin-containing media.
2.5.3 Reduced Vancomycin Susceptibility in S. aureus
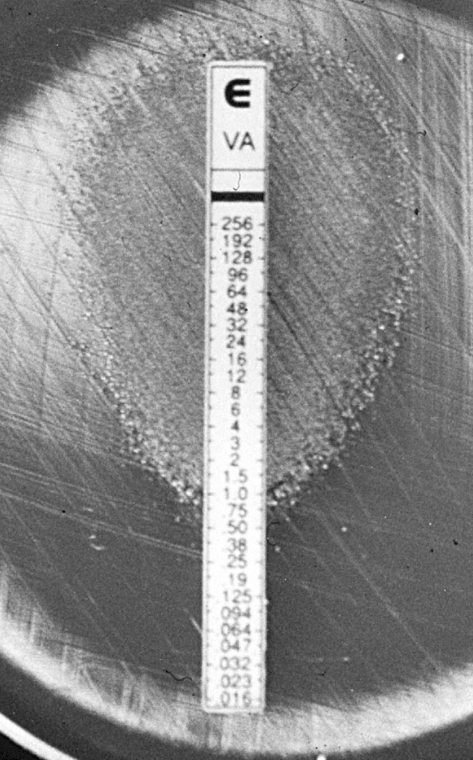
The first MRSA isolate with reduced vancomycin susceptibility was reported by Hiramatsu in 1997 (24). The CLSI breakpoints were lowered in 2006 to reflect clinical failures at MICs of 2 μg/mL; current breakpoints classify S. aureus as susceptible at MIC ≤2, intermediate (VISA) at 4–8, and resistant (VRSA) at ≥16 μg/mL (9).
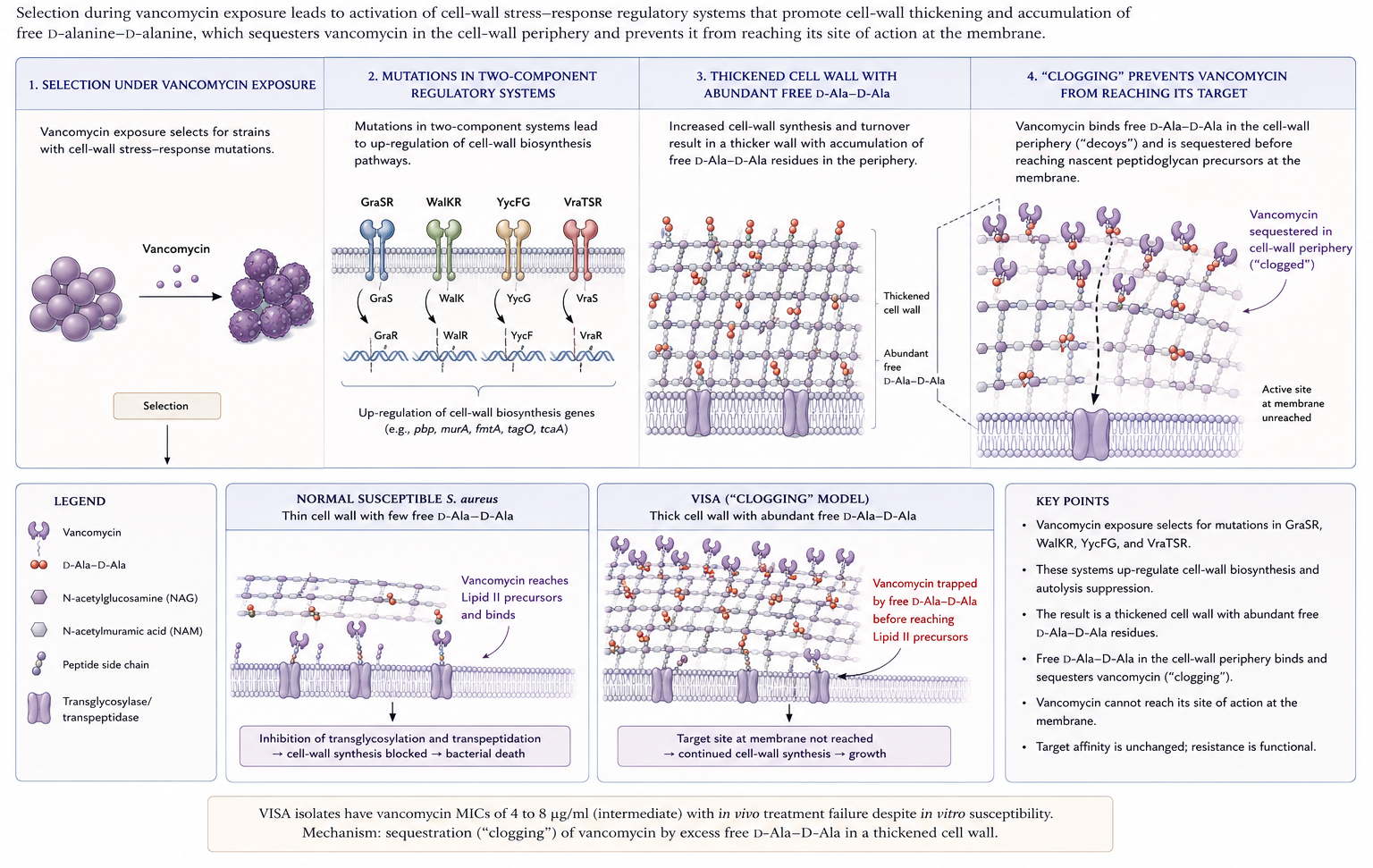
- VISA: MIC 4–8 μg/mL; arises through stepwise mutation under selection pressure; characterised by thickened cell walls with abundant unbound D-Ala-D-Ala residues that “trap” vancomycin in the periphery — the so-called clogging mechanism (Figure 6) (23). The therapeutic implication is that more vancomycin is unlikely to overcome it; switch to alternatives (linezolid, daptomycin, ceftaroline, or a lipoglycopeptide) (26).
- hVISA: heterogeneous VISA — population MIC in the susceptible range (≤2 μg/mL) but containing subpopulations growing at 4–8 μg/mL; precursor of VISA (27,45). Global prevalence ~6% among MRSA in a recent meta-analysis (49); recent reports document hVISA emergence in Latin America (6).
- VRSA: acquisition of the enterococcal vanA operon by MRSA; rare (~16 confirmed U.S. cases by 2023) but high-level resistant (7).
Mutations in two- and three-component regulatory systems (GraSR, WalKR/YycFG, VraTSR) controlling cell-wall homeostasis underlie both hVISA and VISA phenotypes (5,27,28,59).
Bacteraemia caused by MRSA strains with vancomycin MICs at the upper end of susceptibility (1.5–2 μg/mL by Etest) has been linked to higher rates of treatment failure, persistent bacteraemia, and mortality in several observational studies (8,21,38,40,48). A 2014 Cochrane meta-analysis found this association but emphasised the heterogeneity of testing methodologies (19,25,60). IDSA 2011 MRSA guidelines recommend considering an alternative agent for persistent bacteraemia despite adequate vancomycin exposure or for isolates with MIC >2 μg/mL (33).
2.6 Pharmacokinetics and Therapeutic Drug Monitoring
Vancomycin exhibits multi-compartmental kinetics with a distribution half-life of ~30 minutes and an elimination half-life of 6–12 hours in adults with normal renal function. Protein binding is approximately 55%. Tissue penetration is modest into lung epithelial lining fluid, bone, and CSF (with inflamed meninges, CSF levels reach 7%–21% of serum) and very limited into vitreous humour without inflammation.
2.6.1 AUC-guided versus trough-guided dosing
Historically, trough levels of 15–20 mg/L were targeted for serious MRSA infections, based on the surrogate assumption that troughs of this magnitude would achieve an AUC/MIC ratio of ≥400 — the pharmacodynamic threshold derived from pre-clinical and clinical S. aureus data (21,40). However, the relationship between trough and AUC is variable, and trough-guided regimens often expose patients to AUC values >600 mg·h/L — a threshold increasingly associated with acute kidney injury (34).
The 2020 consensus guidelines from ASHP/IDSA/PIDS/SIDP now recommend AUC-guided monitoring by Bayesian software (or first-order pharmacokinetic equations using two timed levels), targeting AUC₂₄ 400–600 mg·h/L for serious MRSA infections (Figure 7) (47). Loading doses of 25–30 mg/kg are suggested for severely ill patients to attain therapeutic exposure earlier.
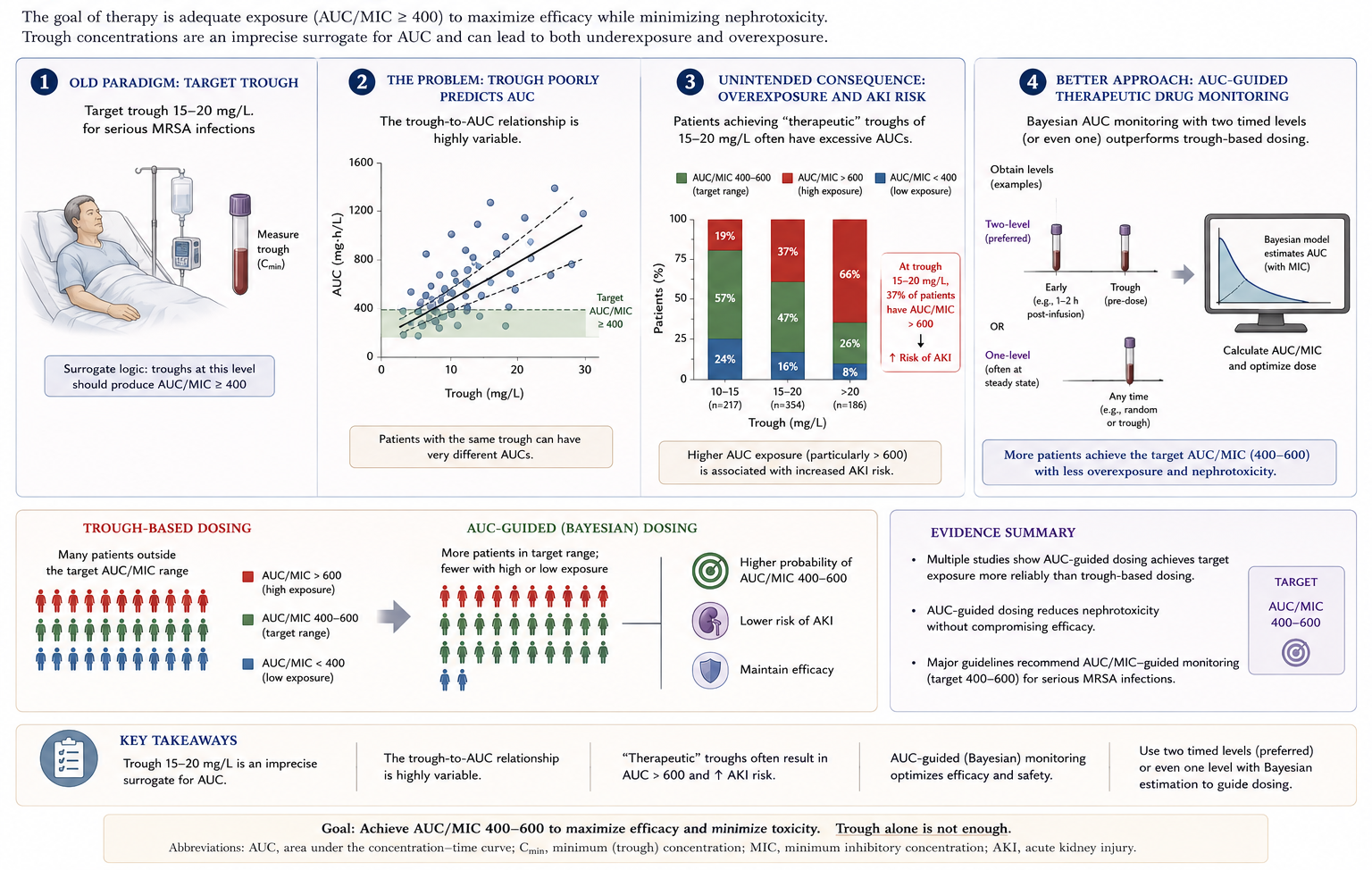
- Average adult dose: 15–20 mg/kg every 8–12 h (actual body weight).
- Loading dose for sepsis/endocarditis/meningitis/pneumonia: 25–30 mg/kg infused at ≤500 mg/h.
- Continuous infusion (30–40 mg/kg/day after loading) may reduce nephrotoxicity without compromising efficacy (20).
- Prospective evidence for clinical benefit of AUC over trough monitoring is still maturing — most supporting evidence is retrospective (47).
2.6.2 Special populations
Hemodialysis: For intermittent HD, the recommended loading dose is 25 mg/kg with maintenance of 7.5–10 mg/kg (low-permeability dialyser) or 10–15 mg/kg (high-permeability dialyser) given during dialysis. CRRT: clearance is significantly higher; 500 mg every 8 h after 20 mg/kg loading is suggested for typical effluent rates (~25 mL/kg/h); higher effluent rates require larger doses (47).
Neonates and children: dosing follows weight- and age-based algorithms; AUC-guided monitoring is recommended where Bayesian tools are available.
Pregnancy: category B/C; minimal transplacental passage in ex vivo models; therapeutic fetal concentrations reached in overt amnionitis.
2.6.3 Local administration
Vancomycin is the preferred agent for intraperitoneal therapy of peritonitis associated with continuous ambulatory peritoneal dialysis (CAPD); 15–30 mg/kg in long dwell (≥6 h) every 5–7 days for intermittent dosing (29). Intraventricular dosing for ventriculitis is 5–20 mg depending on ventricular size and CSF drainage volume; intraocular dosing for endophthalmitis is 1 mg by intravitreal injection.
2.7 Dosing
See Table 1 for a comparative dosing summary across all five agents.
| Drug | Route | Recommended adult dosage | Infusion | Comments |
|---|---|---|---|---|
| Vancomycin | IV | 15–20 mg/kg q8–12h; continuous infusion 30 mg/kg/day after 15 mg/kg loading | ≤5 mg/mL at <15 mg/min (1 g over ≥60 min) | AUC/MIC 400–600 for serious MRSA |
| PO | 125–500 mg q6h | — | C. difficile colitis | |
| Teicoplanin | IV or IM | Loading: uncomplicated MRSA 10 mg/kg q12h × 5 doses OR 12 mg/kg q12h × 3 doses (target 15–30 mg/L); complicated MRSA 12 mg/kg q12h × 5 doses (target 20–40 mg/L); non-MRSA 6 mg/kg q12h × 3 doses. Maintenance 6 mg/kg/day | Bolus permitted | Higher maintenance doses needed for troughs >20 mg/L |
| PO | 100–400 mg q12h | — | C. difficile colitis | |
| Telavancin | IV | 10 mg/kg q24h | 1 h infusion | 25 mg or 750 mg vials reconstituted to ~15 mg/mL |
| Dalbavancin | IV | Two-dose: 1000 mg, then 500 mg one week later. Single-dose: 1500 mg. Paediatric ABSSSI: <6 y 22.5 mg/kg; 6–<18 y 18 mg/kg (max 1500 mg) | 30 min infusion | 500 mg vials in 5% dextrose, 1–5 mg/mL |
| Oritavancin (Orbactiv/Tenkasi) | IV | 1200 mg single dose | 3 h infusion | 400 mg vials in 5% dextrose only, 1.2 mg/mL |
| Oritavancin (Kimyrsa) | IV | 1200 mg single dose | 1 h infusion | 1200 mg vial reconstituted to 4.8 mg/mL |
2.8 Adverse Reactions
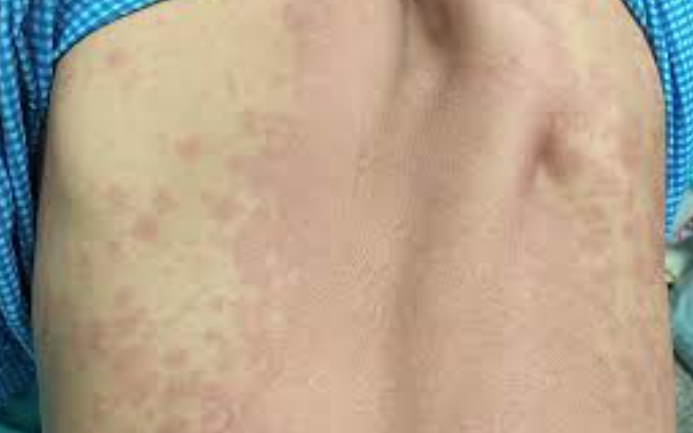
Infusion-related reactions — red-man (red-neck) syndrome — are the most common adverse events with vancomycin, occurring in 3.4%–14% of patients during infusion (Figure 8). The reaction is mediated by direct histamine release from cutaneous mast cells (not IgE-mediated) and is preventable by slowing infusion rate (<10 mg/min) and antihistamine premedication. This is a pseudoallergic reaction, not a true allergy — patients can usually continue vancomycin with slower infusion and antihistamine premedication. True IgE-mediated anaphylaxis to vancomycin is exceedingly rare (42).
Nephrotoxicity has been reported throughout vancomycin’s clinical history. Risk increases with troughs ≥15 mg/L, AUC >650 mg·h/L, concomitant nephrotoxins (especially piperacillin-tazobactam — risk amplified relative to monotherapy or to vancomycin plus cefepime/carbapenem), advanced age, obesity, hypovolaemia, and treatment duration >14 days (21,34). Continuous infusion is associated with lower nephrotoxicity than intermittent dosing in meta-analyses (20).
The increased risk of AKI with the vancomycin + piperacillin-tazobactam combination is consistent across adult and paediatric cohorts and is amplified relative to vancomycin alone, piperacillin-tazobactam alone, or vancomycin combined with cefepime or a carbapenem. Where feasible, consider alternatives (37).
Ototoxicity is uncommon with contemporary vancomycin preparations but can occur — particularly with prolonged courses (>2 weeks) and in very-low-birth-weight infants (39). Effects are usually reversible.
Haematologic: neutropenia in 1%–2% (12%–13% with long-term therapy); thrombocytopenia is rare but may be immune-mediated (vancomycin-dependent platelet-reactive antibodies). Class cross-reactivity with teicoplanin has been described.
Cutaneous and immune-mediated: maculopapular rash (~3%), drug fever (~2%); DRESS, linear IgA bullous dermatosis, and Stevens–Johnson syndrome/TEN are rare but reported.
2.9 Drug Interactions
Vancomycin is incompatible in IV solutions with many compounds including ceftazidime, chloramphenicol, methicillin, corticosteroids, heparin, and sodium bicarbonate. Anion-exchange resins (e.g., cholestyramine) bind oral vancomycin in the gut lumen. Concomitant anaesthetics increase the rate of histamine-release reactions — administer the anaesthetic after the vancomycin infusion.
2.10 Clinical Uses
2.10.1 Skin and soft tissue infections (ABSSSI)
Vancomycin is the comparator in most randomised trials of newer agents against gram-positive ABSSSI; nearly all have demonstrated non-inferiority. Selection among agents depends on local resistance patterns, the possibility of oral step-down or early discharge, tolerability, and cost.
2.10.2 Bacteraemia and infective endocarditis
Vancomycin remains the recommended therapy for MRSA bacteraemia and right-sided endocarditis when daptomycin is unsuitable, despite recognised limitations (slow bactericidal activity, treatment failure with isolates at the upper end of the susceptible MIC range). The IDSA 2011 MRSA guidelines and updated 2025 IDSA bacteraemia guidance recommend AUC-guided dosing with consideration of alternative therapy for persistent bacteraemia ≥7 days despite adequate exposure (30,33).
2.10.2.1 Persistent MRSA bacteraemia — alternatives
When vancomycin fails, the following options are available:
- Daptomycin ≥8 mg/kg (high-dose to reduce resistance emergence)
- Daptomycin + ceftaroline — synergy in vitro; the 2019 Geriak RCT (small but suggestive of survival benefit) has made this standard rescue therapy in many academic centres (17)
- Daptomycin + β-lactam (oxacillin, nafcillin) — the “seesaw effect” where daptomycin susceptibility improves with concomitant β-lactam exposure
- Ceftaroline monotherapy — option for daptomycin-non-susceptible isolates
- Linezolid — bacteriostatic, generally not first choice for bacteraemia but has a role in refractory cases
- Investigational: oritavancin or dalbavancin for off-label bacteraemia/endocarditis continuation
2.10.2.2 VRE bacteraemia — treatment choices
| Agent | Pros | Cons |
|---|---|---|
| Linezolid 600 mg q12h | Oral option; effective; widely available | Bone marrow suppression with prolonged use; serotonin syndrome with SSRIs |
| Daptomycin ≥10 mg/kg | Bactericidal; once-daily | High doses needed; resistance emergence reported |
| Tigecycline | Activity vs VRE | Low serum levels; FDA mortality signal |
| Oritavancin | Single-dose; active vs VanA | Off-label; aPTT interference; cost |
| Quinupristin-dalfopristin | Active vs E. faecium (not faecalis) | IV only; arthralgia/myalgia; venous irritation |
For VRE bacteraemia, daptomycin (high-dose, 10–12 mg/kg) and linezolid are both reasonable first-line choices. Oritavancin is increasingly used off-label — particularly attractive because its long half-life means a single dose can cover much of a typical 14-day course (10,41).
2.10.3 Pneumonia
For hospital-acquired and ventilator-associated MRSA pneumonia, vancomycin and linezolid are recommended first-line; clinical equivalence has been demonstrated in most randomised trials, though linezolid achieves higher pulmonary epithelial lining fluid concentrations (30).
2.10.4 Meningitis and CNS infections
Vancomycin combined with a third-generation cephalosporin is recommended for empirical therapy of bacterial meningitis in adults to cover penicillin-resistant pneumococci, and for CSF shunt–related infections caused by methicillin-resistant staphylococci. Intraventricular dosing supplements systemic therapy when CSF concentrations are inadequate.
2.10.5 Clostridioides difficile colitis
Oral vancomycin (125 mg q6h × 10 days) is recommended for the first episode of non-fulminant C. difficile infection by IDSA/SHEA 2021 guidelines, with fidaxomicin preferred when available. For recurrent disease, tapering and pulsed oral vancomycin regimens or fidaxomicin are appropriate.
3 Teicoplanin
3.1 Chemistry, Spectrum, and Mechanism
Teicoplanin is a complex of five closely related glycopeptide molecules produced by Actinoplanes teichomyceticus, differing from vancomycin by the presence of a fatty-acid side chain that enhances tissue penetration and prolongs half-life. The mechanism of action — D-Ala-D-Ala binding — is identical to vancomycin, and the antimicrobial spectrum largely overlaps, though some staphylococcal species (notably S. haemolyticus) exhibit higher teicoplanin MICs. Activity is preserved against VanB enterococci (which remain teicoplanin-susceptible) but lost against VanA-phenotype enterococci.
3.2 Pharmacokinetics
Teicoplanin’s defining feature is its long elimination half-life (~70–100 h, considerably longer than vancomycin), permitting once-daily maintenance dosing. Protein binding is high (~90%–95%). Tissue penetration is generally better than vancomycin (bone, joint, skin) but CSF penetration remains poor.
3.3 Therapeutic Drug Monitoring
Steady-state trough levels (measured on day 4) correlate with clinical outcome (56). Target troughs are:
- 15–30 mg/L for uncomplicated MRSA infections
- 20–40 mg/L for complicated/severe MRSA infections (endocarditis, bone and joint, deep-seated)
Because of the long half-life, a loading regimen is mandatory to attain therapeutic concentrations promptly — typically 10–12 mg/kg every 12 h for 3–5 doses depending on indication (see Table 1). The loading dose should be administered regardless of creatinine clearance; subsequent maintenance is adjusted for renal function.
A common mistake — omitting or under-loading teicoplanin — produces sub-therapeutic concentrations for days and is associated with treatment failure, particularly in serious staphylococcal infection (56). Always load.
3.4 Clinical Uses
Initial studies using low-dose teicoplanin (3 mg/kg/day) found unacceptably high failure rates (>50%) in severe staphylococcal infection. At higher doses (6–10 mg/kg/day), teicoplanin remained inferior to vancomycin in S. aureus endocarditis and intravascular infection, with failure rates linked to troughs <20 mg/L. Modern dosing with aggressive loading and trough-targeted maintenance produces outcomes comparable to vancomycin for many indications, including ABSSSI, bone and joint infection, and outpatient continuation therapy (53,56).
Teicoplanin is available across Europe, Asia, and South America but not in the United States.
3.5 Adverse Reactions
Teicoplanin is generally less nephrotoxic than vancomycin in meta-analyses (53); the most common adverse events are rash, drug-related fever, and (less commonly) red-neck syndrome. Cross-reactivity for haematologic and hypersensitivity reactions with vancomycin has been described, but is not universal — patients with vancomycin red-neck syndrome or rash often tolerate teicoplanin.
4 Telavancin
4.1 Chemistry and Mechanism
Telavancin is a semi-synthetic lipoglycopeptide derived from vancomycin by addition of a decylaminoethyl side chain and a phosphonomethylaminomethyl group. Like vancomycin it binds D-Ala-D-Ala, but its lipid tail also anchors in the bacterial cytoplasmic membrane, depolarising it and disrupting membrane integrity (Figure 9). This dual mechanism (cell-wall and membrane) produces concentration-dependent rapid bactericidal activity. The lipoglycopeptides modify the parent glycopeptide mechanism with concentration-dependent activity from their membrane component, which is why they tolerate single-dose regimens (22,36).
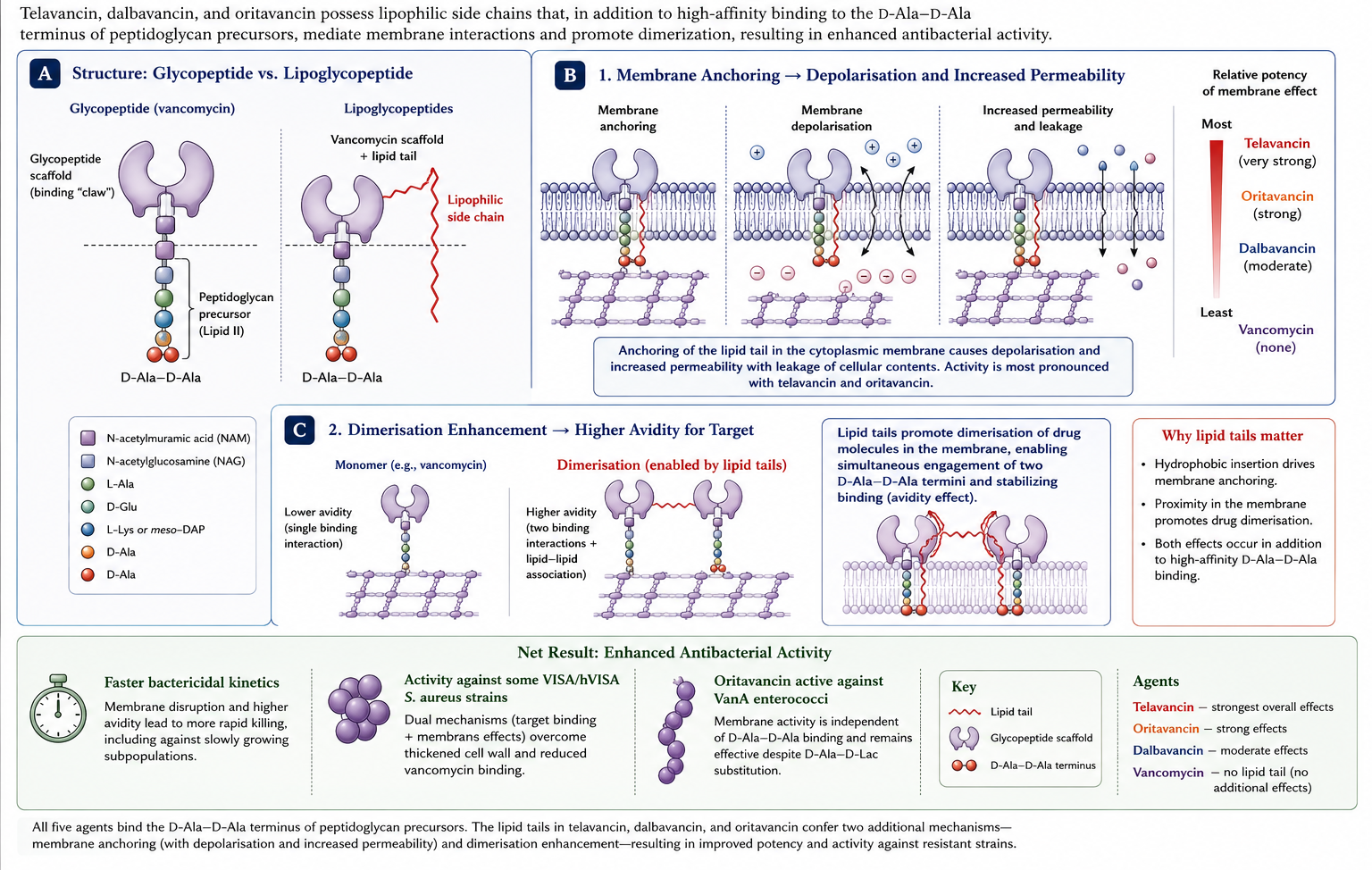
4.2 Antimicrobial Activity
Telavancin is highly active against MRSA (including VISA and hVISA), methicillin-susceptible S. aureus, coagulase-negative staphylococci, streptococci, and vancomycin-susceptible enterococci. MICs are typically 4- to 8-fold lower than vancomycin against susceptible isolates. Telavancin retains activity against most VISA strains but is inactive against VRSA and VanA enterococci.
4.3 Pharmacokinetics
Half-life ~8 h, protein binding ~93%; primarily renal elimination requiring dose adjustment in renal impairment.
4.4 Adverse Reactions
Telavancin carries U.S. FDA boxed warnings for:
- Nephrotoxicity — higher risk than vancomycin, particularly in patients with pre-existing renal dysfunction.
- Fetal risk — animal studies show developmental toxicity; pregnancy testing required before initiation in women of childbearing potential.
Additionally, telavancin causes QTc prolongation — avoid in patients with congenital long-QT syndrome, uncompensated heart failure, severe LV hypertrophy, or concomitant QT-prolonging drugs.
Telavancin artifactually elevates several coagulation assays (PT, aPTT, INR, activated clotting time) by interfering with phospholipid reagents — draw blood for coagulation studies before the next telavancin dose if assessment is needed (51).
4.5 Clinical Uses
Telavancin is approved for:
- Complicated skin and skin-structure infections (cSSSI/ABSSSI) caused by susceptible gram-positive pathogens (10 mg/kg IV daily) — registrational ATLAS trials demonstrated non-inferiority to vancomycin, with cure rates of 88.3% vs 87.1% (51,52).
- Hospital-acquired and ventilator-associated bacterial pneumonia caused by S. aureus when alternative treatments are not suitable — based on the ATTAIN trials, with caveats around the renal-impaired subgroup where mortality favoured vancomycin (46).
5 Dalbavancin
5.1 Chemistry and Mechanism
Dalbavancin is a semi-synthetic lipoglycopeptide derived from a teicoplanin-like A40926 backbone, with a lipophilic side chain that confers a strikingly long elimination half-life (~14 days). Mechanism is D-Ala-D-Ala binding with enhanced dimerisation and membrane anchoring relative to vancomycin.
5.2 Antimicrobial Activity
Dalbavancin is highly active against staphylococci (including MRSA, MSSA, coagulase-negative staphylococci), streptococci, and vancomycin-susceptible enterococci. Activity against VISA is preserved but reduced; VanA enterococci and VRSA are resistant.
5.3 Pharmacokinetics
The extraordinary half-life of ~346 h (~14 days) supports single- or two-dose treatment regimens that complete a full antibiotic course on day 1 or day 8. Protein binding ~93%. Renal clearance is the primary route; no dose adjustment needed for hepatic impairment. Approximately 33% of drug remains in plasma at 14 days.
5.4 Adverse Reactions
Generally well tolerated. The most common adverse events are nausea, headache, diarrhoea, mild ALT elevation, and infusion-related pruritus. CYP450 interactions are minimal.
5.5 Clinical Uses
- ABSSSI: Approved at 1000 mg IV on day 1 followed by 500 mg one week later, OR a single 1500 mg dose (4,13). Non-inferior to comparators (vancomycin → linezolid) in DISCOVER 1 and 2.
- Osteomyelitis: Open-label single-arm trials suggest efficacy for diabetic foot osteomyelitis and native vertebral osteomyelitis at 1500 mg followed by 1500 mg one week later (43).
- Endocarditis and S. aureus bacteraemia: The DOTS trial (Turner 2025) — a randomised controlled trial of dalbavancin (days 1 + 8) versus standard IV therapy for completion treatment of complicated S. aureus bacteraemia after blood-culture clearance — demonstrated noninferiority with comparable safety. This is the first RCT to include right-sided native valve endocarditis (54,55).
Single-dose dalbavancin for ABSSSI can avoid hospitalisation entirely and is increasingly used to facilitate ED discharge or to bridge IV-to-outpatient transitions when adherence with oral step-down is uncertain. Cost remains a barrier in many systems.
5.5.1 DOTS Trial — Population Pharmacokinetics and Exposure–Response
A secondary PK analysis of the DOTS RCT (n = 97, 640 samples) characterised dalbavancin disposition using a 3-compartment population PK model (35):
| Parameter | Estimate | 95% CI | IIV (CV%) |
|---|---|---|---|
| CL | 0.066 L/h | 0.062–0.069 | 22.6% |
| V1 (central) | 5.67 L | 5.37–5.99 | 19.7% |
Protein binding was >99% in 92.3% of paired samples — substantially higher than the ~93% historically estimated from radiolabeled equilibrium dialysis. Albumin was the primary determinant of variability in fraction unbound, not renal function. Key covariate effects included creatinine clearance on CL (exponent 0.21), body weight on all distribution volumes (exponents 0.56–0.82), and albumin on fraction unbound (exponent −0.78) (35).
5.5.1.1 Exposure–response: day 22 concentration
Among 93 evaluable patients, 72 (77.4%) achieved clinical success at day 70. A total day 22 concentration >32 µg/mL was identified as the optimal cut point:
| n | Clinical success | ||
|---|---|---|---|
| C22 >32 µg/mL | 30 | 29 (96.7%) | |
| C22 ≤32 µg/mL | 63 | 43 (68.3%) | |
| Adjusted difference | 25.3 pp | 95% CI 3.5–47.0 |
No increase in serious adverse events was observed in the higher-exposure group. However, mean day 22 concentration with two 1500 mg doses was 29.0 µg/mL — i.e. the majority of patients fall below the 32 µg/mL threshold. 73% of unbound samples were below quantification by day 42, and all infectious complications occurred after day 40, temporally coinciding with declining unbound exposure (35).
The 32 µg/mL threshold requires external validation before clinical application. Exploratory data suggest some patients may benefit from a third dose between days 22–40, guided by low day 22 concentration or applied empirically in patients at risk for lower exposure (high weight, high CrCl, low albumin). This represents a different TDM paradigm from vancomycin — deciding whether to give an additional fixed dose weeks into therapy rather than adjusting a continuous infusion (35).
5.5.2 Dalbavancin in People Who Inject Drugs (PWID)
Dalbavancin is increasingly used for S. aureus bacteraemia and right-sided endocarditis in PWID, where single 1500 mg dosing covers ~2 weeks of therapy without requiring IV access. These patients are at high risk for line infections, often leave against medical advice, and cannot reliably complete a 4–6 week IV course (3,63).
The DOTS trial included right-sided native valve endocarditis and showed dalbavancin to be noninferior to standard IV therapy — the first RCT data covering this population (55). Left-sided and prosthetic valve endocarditis remain supported only by observational data; dedicated trials are needed.
6 Oritavancin
6.1 Chemistry and Mechanism
Oritavancin is a derivative of chloroeremomycin — itself a vancomycin analogue produced by Amycolatopsis orientalis. Like telavancin and dalbavancin, oritavancin has a lipophilic side chain that anchors it in the bacterial membrane, supporting a triple mechanism (Figure 10): inhibition of transglycosylation, inhibition of transpeptidation (through binding of a secondary peptidoglycan site beyond D-Ala-D-Ala), and direct membrane depolarisation. This secondary binding site is what gives oritavancin its activity against VanA enterococci — the cell-wall precursors of VanA enterococci end in D-Ala-D-lactate, which vancomycin cannot bind effectively, but oritavancin can still inhibit transpeptidation through its alternative binding site (2).
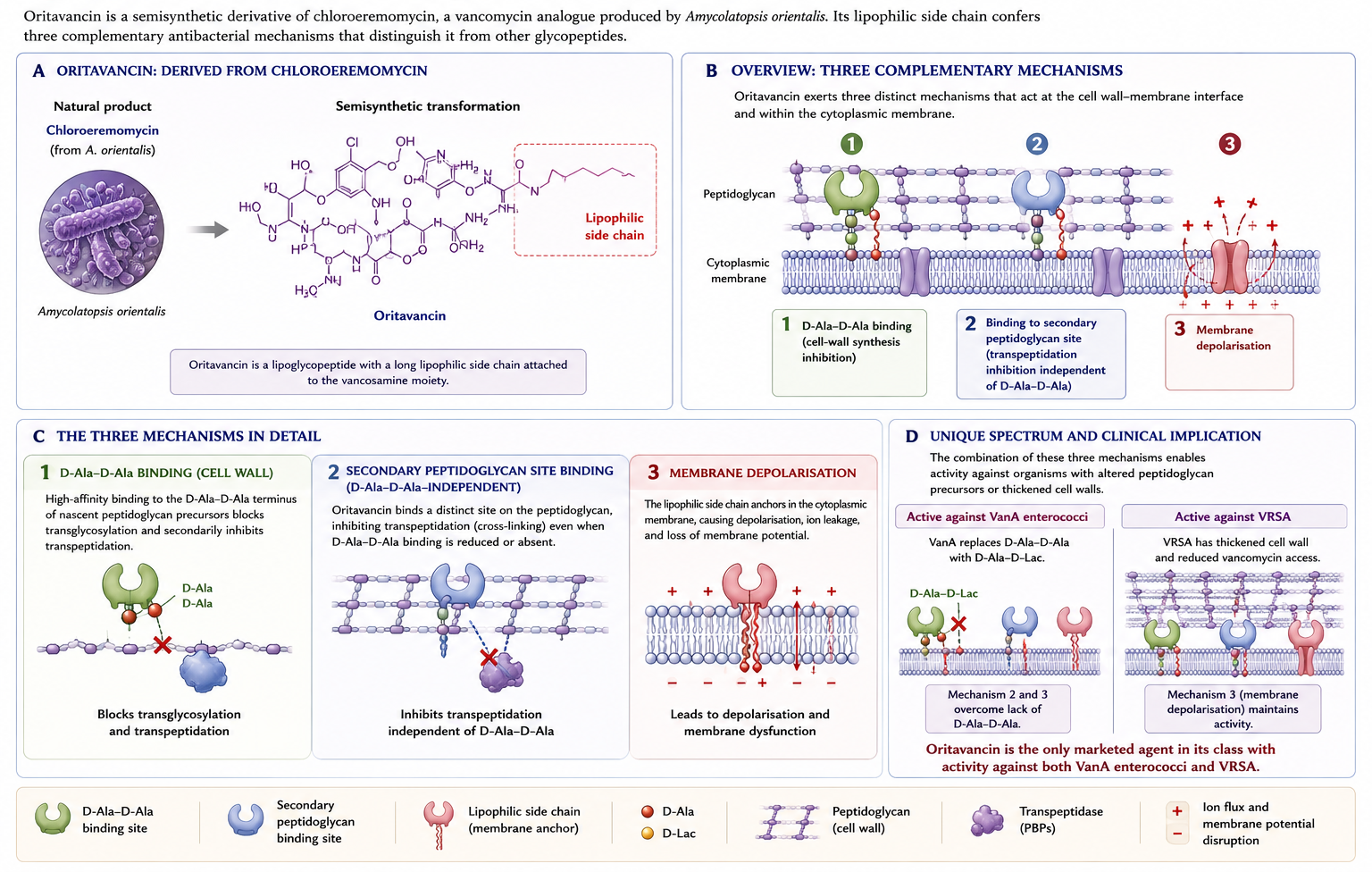
6.2 Antimicrobial Activity
Oritavancin is active against staphylococci (MRSA, VISA, VRSA), streptococci, vancomycin-susceptible and vancomycin-resistant (VanA, VanB) enterococci, and gram-positive anaerobes. Concentration-dependent bactericidal activity.
6.3 Pharmacokinetics
Terminal half-life ~245 h (~10 days). Protein binding ~85%. Long tissue residence allows single-dose dosing for ABSSSI.
6.4 Adverse Reactions and Important Cautions
Oritavancin causes artifactual prolongation of activated partial thromboplastin time (aPTT) for up to 5 days by interfering with phospholipid reagents. Therefore:
- Concurrent IV unfractionated heparin is contraindicated for 5 days after a dose (aPTT cannot be reliably monitored).
- For warfarin monitoring within 24 hours, use a non-aPTT-based method (chromogenic assay).
- PT/INR may also be affected for up to 12 hours; dose adjustments based on aPTT or PT/INR during this period are unreliable.
Other adverse events include headache, nausea, vomiting, diarrhoea, infusion reactions (flushing, pruritus), and mild ALT elevation. Cytochrome P450 interactions are limited but potential — oritavancin is a weak inhibitor of CYP2C9 and CYP2C19 and a weak inducer of CYP3A4 and CYP2D6.
6.5 Clinical Uses
- ABSSSI: Approved at 1200 mg IV as a single dose (Orbactiv/Tenkasi over 3 h, or Kimyrsa over 1 h). Non-inferior to twice-daily vancomycin for 7–10 days in SOLO I and SOLO II (11,12).
- Off-label uses: open-label data describe use for osteomyelitis, prosthetic joint infection, and bacteraemia (57). Single- or multi-dose regimens have been employed.
7 Comparative Summary
| Feature | Vancomycin | Teicoplanin | Telavancin | Dalbavancin | Oritavancin |
|---|---|---|---|---|---|
| Mechanism | D-Ala-D-Ala | D-Ala-D-Ala | D-Ala-D-Ala + membrane | D-Ala-D-Ala + dimerisation | D-Ala-D-Ala + secondary binding + membrane |
| MRSA | ✓ | ✓ | ✓ | ✓ | ✓ |
| VISA | ✓ (reduced) | variable | ✓ | ✓ (reduced) | ✓ |
| VRSA | ✗ | ✗ | ✗ | ✗ | ✓ |
| VanA enterococci | ✗ | ✗ | ✗ | ✗ | ✓ |
| VanB enterococci | ✗ | ✓ | ✗ | ✗ | ✓ |
| Half-life | 6–12 h | 70–100 h | 8 h | ~346 h (~14 d) | ~245 h (~10 d) |
| Renal adjustment | Yes | Yes | Yes | No | No |
| Hepatic adjustment | No | No | No | No | No |
| Pregnancy | Category B/C | Use with caution | Boxed warning | Use only if benefit > risk | Use only if benefit > risk |
| Key AE concern | Nephrotoxicity, red-neck | Less nephrotoxic | Nephrotoxicity, QTc, coag assays | Generally well tolerated | aPTT interference (5 d) |
| US availability | ✓ | ✗ | ✓ | ✓ | ✓ |
8 Stewardship Considerations
The lipoglycopeptides — particularly dalbavancin and oritavancin with their multi-week post-dose tissue persistence — raise important antimicrobial stewardship questions:
- Selection pressure: prolonged sub-MIC tissue concentrations could in principle select for resistance, though this has not been documented at scale in surveillance studies to date (50). Monitoring is warranted.
- Operational use: single-dose ABSSSI therapy can avoid admission, reduce length of stay, and bridge transitions of care — but requires clear protocols for patient selection (excluding deep infection, sepsis, and infections needing source control).
- Cost: acquisition cost is high; cost-effectiveness depends on local hospitalisation costs, the availability of OPAT (outpatient parenteral antimicrobial therapy), and the alternative oral options.
- Off-label use: in osteomyelitis, prosthetic joint infection, and bacteraemia, single-dose regimens are increasingly used, but the evidence base remains predominantly observational. Prospective controlled trials are in progress.
9 Stewardship Decision Framework
9.1 When to use lipoglycopeptides
Strong rationale:
- ABSSSI requiring inpatient gram-positive cover but suitable for early discharge / ED treatment
- Patients with poor adherence to oral step-down or unable to take oral antibiotics
- Difficult IV access where prolonged IV is not feasible
- VRE bacteraemia / VanA enterococcal infection — oritavancin specifically
- Outpatient continuation of osteomyelitis or endocarditis therapy (off-label)
Weaker rationale (think twice):
- Routine ABSSSI in patients who can take oral cephalexin/clindamycin/doxycycline
- Hospital-acquired infections where source control is not established
- Patients with concurrent need for IV unfractionated heparin (avoid oritavancin)
9.2 Decision framework — empirical gram-positive cover
| Scenario | First-line | Lipoglycopeptide role |
|---|---|---|
| ABSSSI, inpatient, oral step-down possible | Vancomycin → oral step-down | Single-dose dalbavancin if ED discharge desired |
| ABSSSI, ED, avoid admission | — | Single-dose dalbavancin or oritavancin |
| MRSA bacteraemia, complicated | Vancomycin (AUC) or daptomycin | Dalbavancin for continuation (off-label) |
| MRSA endocarditis | Vancomycin or daptomycin (+ ceftaroline) | Investigational role |
| HAP/VAP, MRSA | Vancomycin or linezolid | Telavancin only if alternatives unsuitable |
| Diabetic foot osteomyelitis, MRSA | Standard regimen | Dalbavancin two-dose attractive |
| VanA VRE bacteraemia | Linezolid, daptomycin (high-dose), tigecycline | Oritavancin if other options exhausted |
| C. difficile colitis | Oral vancomycin or fidaxomicin | No role |
9.3 Pharmacoeconomics
The cost-effectiveness of lipoglycopeptides depends on local hospitalisation costs:
- Acquisition cost: dalbavancin €1,500–2,500 per 1500 mg dose; oritavancin €2,500–3,500 per 1200 mg dose (varies by country)
- Per-day hospital ward cost: €500–1,500 in most European systems
- Breakeven: approximately 2–3 hospital days avoided per single-dose lipoglycopeptide
- Cost-effective when avoiding admission entirely for ABSSSI, discharging early from bacteraemia, or replacing a prolonged OPAT IV course
- Less cost-effective when displacing inexpensive oral options
10 Clinical Cases for Discussion
10.1 Case 1 — MRSA bacteraemia with rising creatinine
A 55-year-old man with type 2 diabetes is admitted with right-sided infective endocarditis and MRSA bacteraemia. Day 5 of vancomycin (trough 18 mg/L, AUC 580). Creatinine has risen from 0.9 → 1.6 mg/dL. Blood cultures remain positive at 96 hours.
Key considerations:
- The persistent bacteraemia at 96 hours is the critical signal — IDSA recommends considering alternative therapy for persistent bacteraemia ≥7 days despite adequate vancomycin exposure
- The rising creatinine adds urgency to switching agents
- Imaging (TEE, CT for metastatic foci) to look for uncontrolled source
- Switch to daptomycin 10 mg/kg ± ceftaroline for synergy/seesaw effect
- Discontinue concomitant nephrotoxins
10.2 Case 2 — ABSSSI in IV drug user
A 32-year-old woman, active opioid injection use, presents with extensive cellulitis at an injection site. Stable, no systemic features. MRSA likely. Refusing admission, no IV access available, unreliable for outpatient appointments.
Key considerations:
- This is the textbook indication for single-dose lipoglycopeptide therapy
- Either dalbavancin (1500 mg, 30 min infusion) or oritavancin (1200 mg) would be reasonable
- Dalbavancin’s shorter infusion time is operationally easier in the ED
- Cost-effectiveness is generally favourable when admission would otherwise be needed
- Structured post-treatment follow-up by phone or community health workers is recommended
10.3 Case 3 — VanA VRE bacteraemia
A 68-year-old patient, post-cardiac surgery in ICU day 21, develops new fever with positive blood cultures: E. faecium VanA phenotype. The patient has acute kidney injury (CrCl 30 mL/min) and is on dual antiplatelet therapy.
Key considerations:
- Linezolid is a strong first-line choice: 100% oral bioavailability, no renal adjustment, good VRE evidence
- Daptomycin (high-dose) works but renal impairment limits dose escalation
- Oritavancin is increasingly attractive (single dose covers a 14-day course) but the aPTT interference complicates anticoagulation monitoring
- Tigecycline should generally be avoided for bacteraemia given the FDA mortality signal
- Monitor for bone marrow suppression if linezolid is used beyond 14 days
11 Conclusions
Vancomycin remains the workhorse for serious gram-positive infection, with AUC-guided dosing now standard for serious MRSA infection. Teicoplanin offers an attractive alternative in regions where it is available, with possibly lower nephrotoxicity and once-daily dosing. The lipoglycopeptides — telavancin, dalbavancin, oritavancin — have expanded the therapeutic options, with dalbavancin and oritavancin in particular offering single- or two-dose regimens uniquely suited to ABSSSI and selected off-label indications. The high cost of these newer agents and the need for prospective evidence in serious off-label uses limit broader deployment.